Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
[Special Issue] Exercise and cardioprotection against ischemia reperfusion injury: A Review
Time:2020-05-08
Number:11129
Author Affiliations

Conditioning Medicine 2020. 3(2):59-70.
Abstract
Cardiovascular disease (CVD) has been the leading cause of death in the United States for nearly a century. Despite relatively recent progress in cardiovascular health interventions, CVD and particularly ischemic heart disease remain a significant global health problem. Various approaches to evoke a preconditioned phenotype has largely been unsuccessful, highlighting several logistical problems in pharmacologic approaches to cardioprotection. Issues include the unpredictable nature of ischemic events, the cost of chronically delivering pharmacologic countermeasures, and the fact that many exogenous stimuli to cardiac preconditioning result in rapid cellular habituation and a waning of cardioprotection. Accordingly, many lifestyle approaches to cardiac preconditioning have been investigated, including moderate intensity aerobic exercise. Regular exercise represents a pragmatic, sustainable, and cost-effective measure for reducing CVD risk and for provoking robust levels of cardioprotection against ischemic events. The scientific sub-field borne from this research line is known as exercise-induced cardiac preconditioning. Key discoveries of the nature of exercise preconditioning reveal that clinically relevant protection can be obtained with moderate intensity exercise in as little as 1-3 days and occurs independent of sex and age. The short-term nature of the exercise preconditioning stimulus highlights the importance of biochemical alterations as a key mechanistic process for this protection. Importantly, mechanisms central to exercise-induced cardiac preconditioning include a concert of up-regulated, or allosterically altered, factors that mitigate calcium overload, free radical damage and oxidative stress, and bioenergetic dyshomeostasis. Finally, the efficacy of exercise-induced cardiac preconditioning highlights the value of repetitive physical activity as a scientific approach to drug discovery.
Keywords: exercise, cardioprotection; ischemia-reperfusion injury; preconditioning; ischemic heart disease
Abstract
Cardiovascular disease (CVD) has been the leading cause of death in the United States for nearly a century. Despite relatively recent progress in cardiovascular health interventions, CVD and particularly ischemic heart disease remain a significant global health problem. Various approaches to evoke a preconditioned phenotype has largely been unsuccessful, highlighting several logistical problems in pharmacologic approaches to cardioprotection. Issues include the unpredictable nature of ischemic events, the cost of chronically delivering pharmacologic countermeasures, and the fact that many exogenous stimuli to cardiac preconditioning result in rapid cellular habituation and a waning of cardioprotection. Accordingly, many lifestyle approaches to cardiac preconditioning have been investigated, including moderate intensity aerobic exercise. Regular exercise represents a pragmatic, sustainable, and cost-effective measure for reducing CVD risk and for provoking robust levels of cardioprotection against ischemic events. The scientific sub-field borne from this research line is known as exercise-induced cardiac preconditioning. Key discoveries of the nature of exercise preconditioning reveal that clinically relevant protection can be obtained with moderate intensity exercise in as little as 1-3 days and occurs independent of sex and age. The short-term nature of the exercise preconditioning stimulus highlights the importance of biochemical alterations as a key mechanistic process for this protection. Importantly, mechanisms central to exercise-induced cardiac preconditioning include a concert of up-regulated, or allosterically altered, factors that mitigate calcium overload, free radical damage and oxidative stress, and bioenergetic dyshomeostasis. Finally, the efficacy of exercise-induced cardiac preconditioning highlights the value of repetitive physical activity as a scientific approach to drug discovery.
Keywords: exercise, cardioprotection; ischemia-reperfusion injury; preconditioning; ischemic heart disease
1. Introduction
Cardiovascular disease (CVD), including ischemic heart disease, remains a leading form of morbidity and mortality across the world (Virani et al., 2020). In response, scientists continue to search for counter measures to CVD that are simultaneously pragmatic, cost effective, and sustainable. One such approach includes the decades-long attempt to reverse engineer preclinical understanding of biochemical cardioprotection into a pharmacological counter therapy against CVD insults. Efforts to date, however, have been thwarted by the faceted and evolutionary nature of CVD, the inability to prognosticate ischemic events, and a Quixotic search for an exogenous pharmacologic agent that will produce lasting cardioprotection without cellular habituation (Bolli and Marban, 1999; Quindry and Franklin, 2018). In the absence of a viable pharmacologic solution to counter CVD incidence and severity, physicians and scientists have also explored lifestyle interventions, including a cardiovascular exercise regimen. Indeed, exercise and other healthy lifestyle counter therapies precondition the myocardium against a host of pathological stressors, including ischemia.
The cardioprotective nature of aerobic exercise is well-established (Paffenbarger et al., 1966; Powers et al., 1998; Starnes et al., 2003). Moreover, while structured exercise (e.g. 3-5 days/week, 30-60 minutes in duration, at an intensity of 50%-85% heart rate reserve) clearly improves cardiovascular health and preconditions the heart against CVD (Garber et al., 2011), accumulated amounts of less regimented physical activity are also protective (Riebe et al., 2018). Initial evidence in this regard was observed more than half a century ago from epidemiological studies, which demonstrated a relationship between occupational physical activity and cardiovascular disease outcomes (Paffenbarger et al., 1966). Relevant to the current discussion, this early epidemiological study of cardioprotection provided an almost prescient conclusion (Paffenbarger et al., 1966):
“The association between work activity and coronary mortality, when considered with the lack of such association with stroke mortality, suggests that physical activity influences the myocardium or its function more than the atherosclerotic process.”
Indeed, the capacity of repetitive muscular work (formal exercise or plentiful physical activity) to induce biochemical cardiac preconditioning was so powerful that it was apparent to epidemiologists of the mid 20th century.
Over the past few decades, much has been uncovered about the mechanisms of this protection. In addition to decreasing risk factors associated with the development of cardiovascular diseases, regular exercise directly protects against cardiac ischemia- reperfusion (IR) injury via cellular biochemical alterations (Demirel et al., 2001; Lennon et al., 2004a; Quindry et al., 2005; Quindry et al., 2012). Thus, given the fact that exercise can precondition the myocardium indefinitely, recent scientific interest has been directed at uncovering mechanisms of exercise cardioprotection for the purpose of designing pharmaceuticals aimed at mitigating damage from IR injury. As is the case in many applied lines of biomedical research, much of the fundamental knowledge about exercise induced cardioprotection against CVD is derived from animal studies, primarily using rodent models. Moreover, while exercise improves outcomes in models of heart failure, vascular dysfunction, and metabolic disease, the largest body of work relates to ischemic heart disease (Powers et al., 1998; Demirel et al., 2001; Starnes et al., 2003; Lennon et al., 2004a; Quindry et al., 2005; Quindry et al., 2012). Accordingly, the thrust of this review is directed toward the latter, primarily detailing what is known about exercise preconditioning against myocardial infarction. Moreover, the topic of exercise induced cardioprotection has been reviewed elsewhere with an emphasis toward scientific understanding of signaling pathways (Starnes and Taylor, 2007; Penna et al., 2020), as a unique scientific approach (Brown and Moore, 2007; Powers et al., 2014; Quindry, 2017; Wojcik et al., 2018), and as an integrated pharmacologic and rehabilitative approach to the treatment and prevention of cardiovascular disease (Fiuza-Luces et al., 2018; Quindry and Franklin, 2018; Thijssen et al., 2018; Chowdhury et al., 2019). The current review updates these perspectives within the more holistic narrative of exercise as a potent means of conditioning medicine for improved heart health in those at significant risk for an ischemic event.
Experimental paradigms of exercise preconditioning against myocardial ischemia
Animal models used to investigate exercise induced cardioprotection, commonly performed in rats and mice, include various exercise protocols followed by surgically induced IR in vivo, or global ischemia applied ex vivo. Although protocols have varied in both the exercise volume employed and duration of IR investigated, overall, many studies have employed exercise volumes comparable to human exercise capacity and measurement of clinically relevant outcomes observed in humans. Table 1 provides a description of a common study design for experimental models of exercise induced cardioprotection. The reader is further directed to a recent ‘systematic comparison of exercise training protocols on animal models of cardiovascular capacity’ for a more in depth presentation of exercise protocols (Feng et al., 2019).
Table 1. Methodological approach common to exercise-induced cardioprotection research studies using rodents and other mammals.
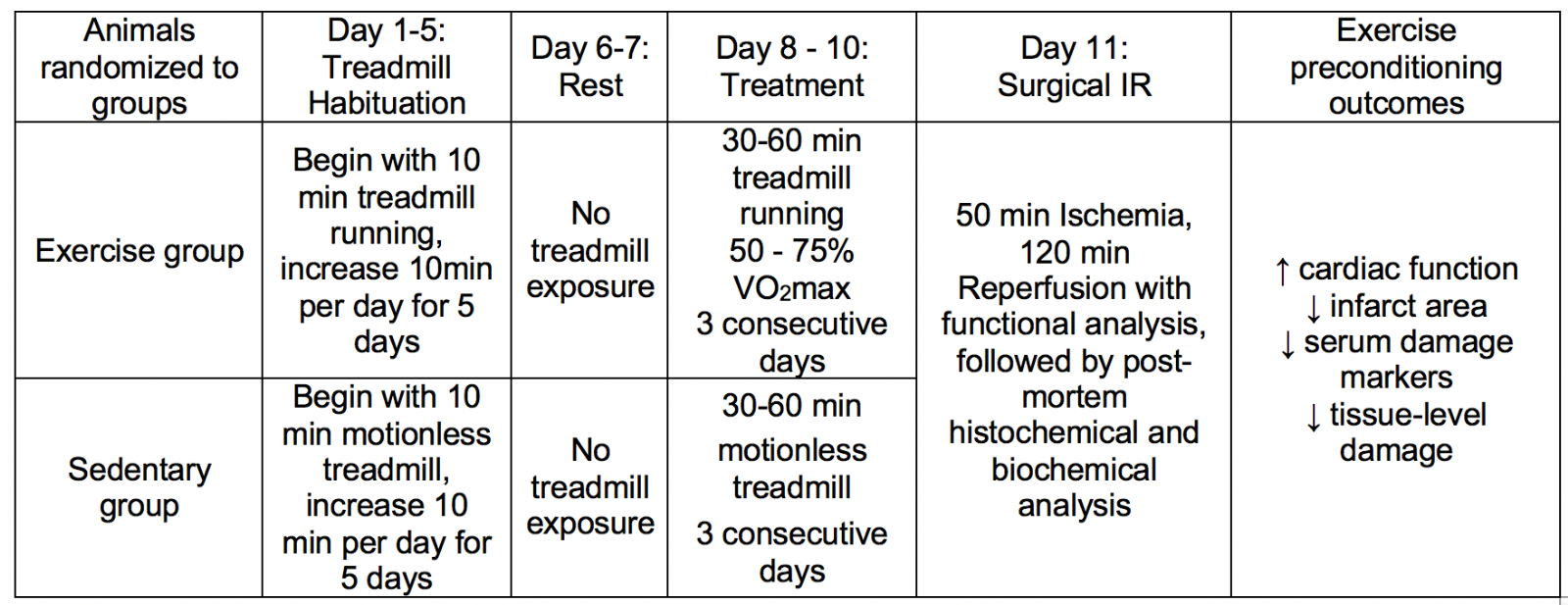
Scientific efficacy of these preclinical studies, of course, is reliant upon the applicability of the exercise interventions in addition to the clinical relevance of the infarct challenge and key dependent outcome measures of cardiac pathology. To this end, it is essential to recognize that the exercise prescriptions used for improving cardiovascular health in humans are proportionally scaled to the animal model of interest. Specifically, the American College of Sports Medicine 10th edition of Guidelines for Exercise Testing and Prescription can be redefined for exercise frequency, intensity, duration, modality, and progression in animal studies (Riebe et al., 2018). Indeed, the exercise frequency of 3-5 days/week of aerobic exercise is common to both human exercise prescriptions and rodent exercise models (Criswell et al., 1993; Powers et al., 1998; McGinnis et al., 2015; Riebe et al., 2018). Accordingly, while the modes of continuous aerobic exercise involve forced treadmill exercise (typically with an electrified shock grid on the tail end of the running surface) or immersive swimming, the modalities do parallel human physical activity. Similarly, the metabolic costs of these activities can be accurately quantified in both humans and animals using applications of indirect calorimetry (Lawler et al., 1993; Riebe et al., 2018). Based on this understanding, the exercise intensity prescribed in exercise-induced cardioprotection studies spans the identical intensity window used in humans (Bowles et al., 1992; Starnes et al., 2003; Lennon et al., 2004a; Riebe et al., 2018).
It is worth acknowledging that forced exercise in rodent models is a potentially confounding aspect of most exercise-induced cardioprotection studies (Mancardi et al., 2009). Fortunately, however, evidence from investigations of rodent-selected wheel running support the application of human exercise prescriptions in animals, noting that when mice are provided the chance to exercise at their volition on a running wheel, they will naturally accumulate a volume of running that equals the prescribed distance outlined in experimental models of exercise (Lightfoot et al., 2010). In aggregate, these findings provide validation to the common exercise approaches used to evoke a cardioprotective phenotype against ischemic insults.
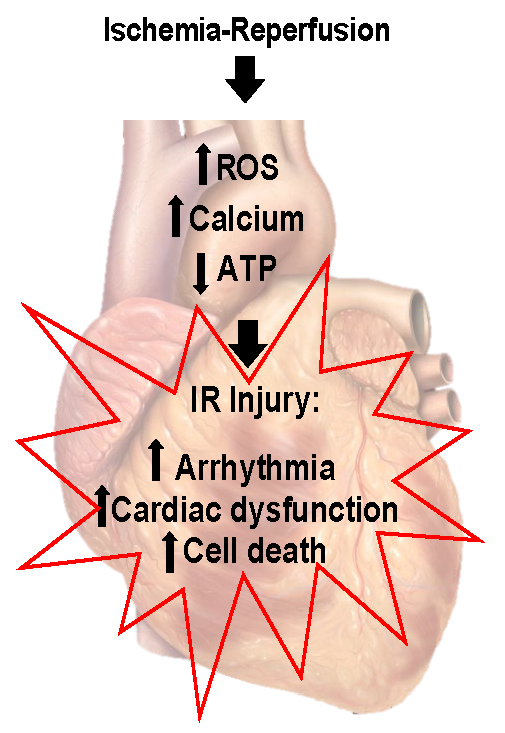
As mentioned previously, clinically relevant outcomes and key dependent measures are essential to understanding the role of exercise and an ischemic-resistant heart. An overview of the pathology of IR has been summarized in Figure 1. Given that myocardial ischemia results in an evolutionary pathology, common clinical endpoints are needed. Clinically relevant outcomes can be most comprehensively examined in surgical ligation models in vivo. In these studies, electrocardiogram (ECG) analysis is readily quantified during both ischemia and reperfusion, and ventricular ectopy can be tracked for potentially lethal arrhythmias using clinical scoring systems based on Lambeth convention dictates (Curtis and Walker, 1988; Miller et al., 2012). Similarly, ventricular pump function can be precisely quantified by pulse pressure measurements from indwelling arterial pressure catheters, pressure volume loop analysis, transthoracic echocardiography, and isolated perfused working heart preparations (Hamilton et al., 2001; Quindry et al., 2005; Starnes et al., 2005). Perhaps most commonly examined, infarct size can be accurately quantified by examining the release of cardiac-specific enzymes from blood plasma or cardiac perfusate collected from isolated heart preparations (Hamilton et al., 2003; Lennon et al., 2004a). In addition, post-mortem histological cross-sections can be used to track both the ischemic area and the infarct region produced by surgical ligation of the left anterior descending artery (LAD) (Hamilton et al., 2003; Quindry et al., 2007; Quindry et al., 2012). Finally, post-mortem examination of the ventricular muscle can be examined for biochemical alterations such that the ischemic area can be compared to perfused regions of the same heart. This simple, but powerful, scientific approach provides invaluable insights to the biochemical alterations in hearts from exercised and sedentary treatments (Quindry et al., 2010a; McGinnis et al., 2015; Miller et al., 2015). As will be discussed subsequently, this approach to using each heart as its own control has been foundational in the early studies to suggest that exercise training appears to cardioprotect through subtle modifications in cellular processes such as apoptosis and autophagy (Quindry et al., 2005; Quindry et al., 2007; Quindry et al., 2012; McGinnis et al., 2015; Miller et al., 2015).
In a new window | Download PPT
Figure 1: Ischemia reperfusion injury. Ischemia-reperfusion results in arrhythmia, decreased cardiac function, and cell death. Elevated free radical production, calcium overload, and decreased mitochondrial function are central to ischemia-reperfusion injury. Figure created by modification of an image created by Patrick J. Lynch, medical illustrator; C. Carl Jaffe, MD, cardiologist. https://creativecommons.org/licenses/by/2.5/
Efficacy in various populations and required exercise dose
Descriptive evidence supporting the notion of exercise-induced cardiac preconditioning has accumulated over the last 25-30 years. Foundational to this line of work is the question, is exercise induced cardiac preconditioning effective for both sexes and across the lifespan? Numerous studies confirm that both males and females exhibit equally robust protection against infarction (Brown et al., 2005a; Chicco et al., 2007). Perhaps most important, given that CVD accrues as a function of advancing age, is evidence from rodent models that exercise induced cardioprotection is effective against ischemic insults in both young and old animals (Starnes et al., 2003; Quindry et al., 2005). Thus, these early studies confirm that exercise induced cardioprotection can be conferred in essentially all hearts, apparently without exclusion based on age or sex.
When it comes to prescribing exercise to prevent or treat CVD, the frequency, type, dose, and intensity of exercise are all important. As mentioned earlier, the methodological approach to exercise training in rodents (and larger mammals) is proportional to exercise prescriptions used to produce improved health and fitness in humans. Accordingly, one would be inclined to ask, what is the dose of exercise needed to produce a cardioprotected phenotype?
In regard to the dose of exercise needed to produce a cardioprotected phenotype, long-term, regular exercise clearly promotes an improved risk factor profile and beneficial alterations in vascularization and structural improvements in ventricular architecture (Gielen et al., 2015). Likewise, regular exercise performed over the course of many weeks results in improved cardiac function and decreased damage and infarct area following IR (Bowles et al., 1992; Powers et al., 1998; Brown et al., 2005b). Interestingly, subsequent investigations demonstrate protection is achieved in as little as 1-3 days of exercise (Demirel et al., 2001; Brown et al., 2005a). That is, exercise preconditioning against ventricular dysrhythmias, decrements in ventricular pump function, and infarct damage occur with just 1-3 daily bouts of exercise prior to IR (Yamashita et al., 1999; Starnes et al., 2003; Taylor et al., 2007; Quindry et al., 2010a;Quindry et al., 2012). Indeed, there is little to no conflicting evidence that short-term bouts (days) of exercise training are sufficient to produce a robustly protected phenotype. Moreover, while the preconditioned state may be achieved within a few days, it important to point out that the protected phenotype is by all accounts sustained with continued participation in exercise (Powers et al., 1998). This finding, while perhaps obvious to some readers, starkly contrasts non-exercise approaches to cardiac preconditioning where cellular habituation of the underlying mechanisms occurs within days (Lefer and Bolli, 2011; Peart et al., 2011).
Quite interestingly, both moderate and high intensity exercise are protective, with the duration of many studies set at 30-60 min per day (Starnes et al., 2003; Lennon et al., 2004a; Quindry et al., 2012; Miller et al., 2015). As discussed earlier, many studies have used exercise interventions that mirror the current baseline physical activity recommendations for adults according to the American College of Sports Medicine, which is moderate intensity exercise for 30-60 min per day 3-5 days per week (Garber et al., 2011; Riebe et al., 2018). This finding is important because it suggests that the ‘dose’ of exercise is relatively low compared to maximal capacity in humans and that the level of exercise required for cardioprotective benefit is obtainable in various populations/fitness levels. It appears that moderate and high intensity exercise are equally protective (Lennon et al., 2004a) though some evidence indicates there may be a slightly greater level of protection regarding post-ischemic cardiac function with higher intensity exercise (Bowles et al., 1992). Follow up studies are needed to further characterize the effect of intensity on cardioprotective outcomes. Additionally, although most studies have employed moderate intensity aerobic exercise, it appears resistance exercise may also confer cardioprotective benefit (Soufi et al., 2011), although more evidence is needed due to the limited number of studies on this topic. Additionally, the protective nature of acute exercise appears to be threshold dependent, where longer exercise bouts do not necessarily translate to greater protection (Lennon et al., 2004a). The relatively short time-course needed to obtain clinically relevant protection underscores the notion that the primary mechanisms of exercise cardiac preconditioning are biochemical in nature and can occur independent of cardiac remodeling.
Limitations to animal models of exercise induced cardiac preconditioning
Perhaps needless to state, animal models of exercise induced cardiac preconditioning allow for invasive measurements that would be difficult and/or impossible in human populations. Despite the clear benefits animal models provide, it is important to note significant inherent limitations. Many studies use rodents that are free from CVD prior to surgically induced IR, which makes direct comparisons to the human population where CVD is present difficult. Although support exists for the presence of exercise induced cardiac preconditioning in humans, as will be discussed in a later section, it must be acknowledged that the healthy heart may respond differently to an exercise stress as compared to an unhealthy heart. However, some studies have investigated exercise induced cardiac preconditioning in disease models. For example, exercise is cardioprotective against the progression of heart failure in an animal model of left ventricular hypertrophy (Marshall et al., 2013). Additionally, exercise remains effective at conferring protection in the presence of traditional risk factors for CVD such as obesity, elevated blood glucose, insulin, and cholesterol levels (Pons et al., 2013; Lund et al., 2015). Finally, co-morbidity disease models, combined with a surgically induced ischemic challenge, confirm the potent role of exercise to cardioprotect despite the presence of uncontrolled diabetes (Kupai et al., 2015).
Although exercise appears to be equally cardioprotective in male and females, mechanistic differences may exist (Brown et al., 2005a; Chicco et al., 2007). While many studies have used female animals or both male and female animals, a large portion of studies have used only male animals, therefore it is reasonable to hypothesize that undiscovered mechanistic differences between sexes likely remain. Lastly, as CVD and ischemic heart disease increase with advancing age, investigations including aged animal models are necessary. Although evidence suggests exercise induced cardiac preconditioning remains effective in older animals (Starnes et al., 2003; Quindry et al., 2005), many studies have not used older animals. Moreover, it cannot be overlooked that in the 2005 investigation by Quindry and colleagues, where TUNEL staining was used as a surrogate of apoptotic processes in the heart, the level of TUNEL-positive nuclei in unstressed aged animals was elevated in an age-dependent fashion. While the exercise intervention was short-term (days), there is no reason to believe that long-term exercise training would have completely mitigated the age-dependent apoptotic process in the heart (Quindry et al., 2005). In total, it is encouraging to note that exercise acutely preconditions the aged heart, but that in the context of conditioning medicine, where most individuals are not life-long exercise habituated, the intervention is not likely to reverse decades of aging and disuse. Indeed, this fact underpins the modern call to action to investigate exercise preconditioning in aged animal models.
Mechanisms of exercise induced cardioprotection against an ischemic insult
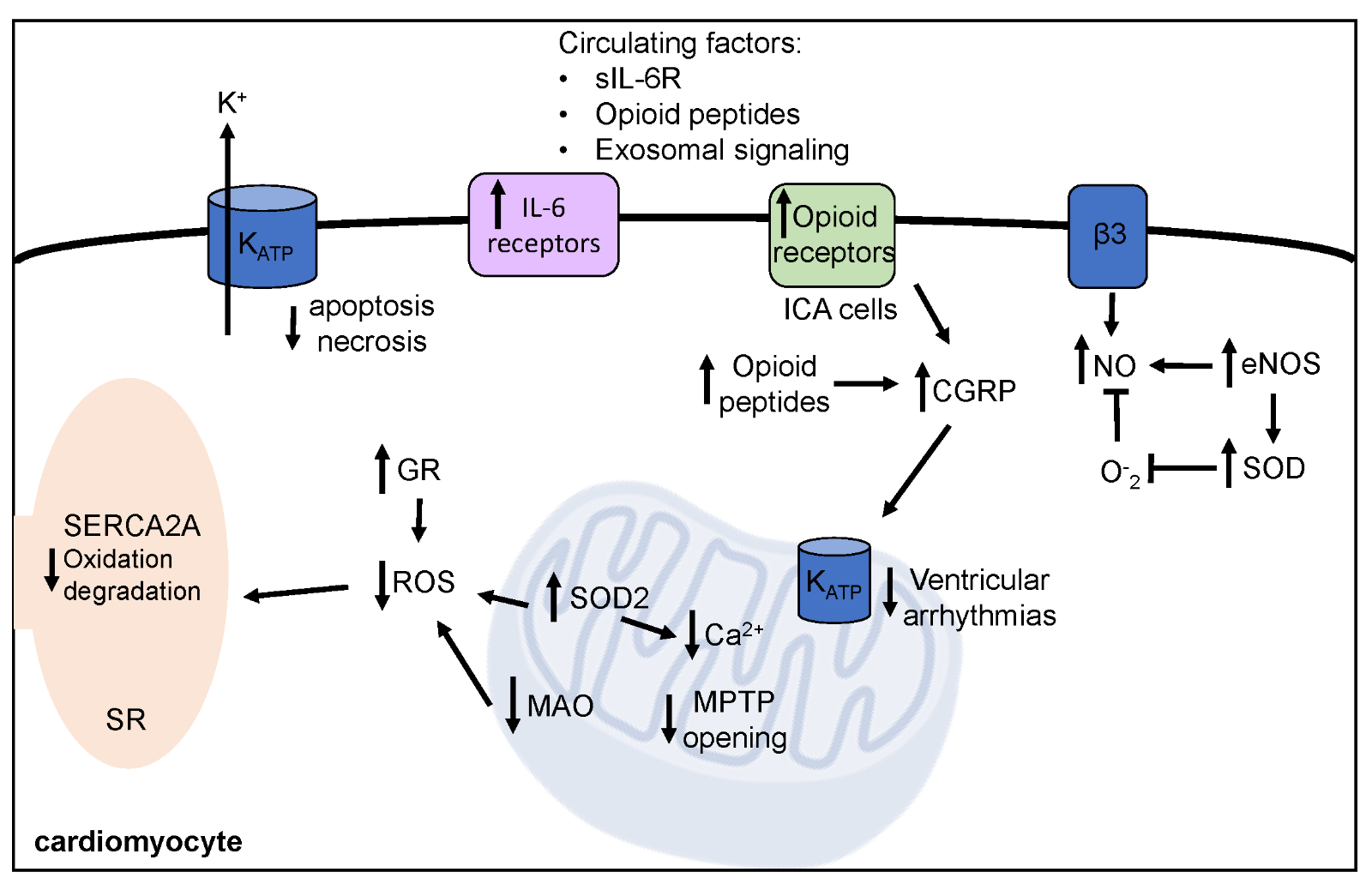
It is well established that acute, (1-3 days) moderate intensity exercise improves outcomes following IR. Specifically, exercised hearts maintain greater function by preserved contractility (Starnes et al., 2003; Lennon et al., 2004a; Quindry et al., 2005), better maintenance of systolic blood pressure (Demirel et al., 2001; Hamilton et al., 2003), and decreased occurrence of arrhythmias (Quindry et al., 2010a; Miller et al., 2012; McGinnis et al., 2015). Exercise preconditioning also results in smaller infarct area, decreased levels of apoptotic cell death, and circulating markers of cardiac tissue damage (Lennon et al., 2004a; Quindry et al., 2007; McGinnis et al., 2015; Miller et al., 2015). Given the short-term nature of the robust protection afforded by exercise preconditioning, biochemical mechanisms that quickly respond to the exercise stimulus are of particular interest and include alterations in antioxidant capacity, ion channel function, and various signaling molecules and pathways. Current understanding of these mechanisms is reviewed in the following sections with a summary provided in Figure 2.
In a new window | Download PPT
Figure 2: Mechanisms of exercise induced cardioprotection against ischemia-reperfusion injury. Exercise prior to ischemia-reperfusion decreases arrhythmia, improves cardiac function, and decreases cell death. Short-term exercise induces protective biochemical alterations within the cardiomyocyte, which are summarized here. SOD2, mitochondrial isoform of superoxide dismutase; GR, glutathione reductase; MAO, monoamine oxidase; MPTP, mitochondrial permeability transition pore; NO, nitric oxide; eNOS, endothelial nitric oxide synthase; SERCA2A, sarcoplasmic endoplasmic calcium ATPase; KATP, ATP-sensitive potassium channels; IL-6, interleukin-6, sIL-6R, soluble IL-6 receptor; ICA, intrinsic cardiac adrenergic; CGRP, reticulum calcitonin gene related peptide.
Antioxidants
Increased free radical production and elevated intracellular calcium levels occur during myocardial IR, resulting in cellular dysfunction and damage (Zweier, 1988; Grill et al., 1992). Limiting the production of free radicals and resulting oxidative stress protects against cell death and dysfunction during IR (Grill et al., 1992). Exercise is well documented to result in upregulated antioxidant defenses, which represents a primary mechanism for limiting cell dysfunction and death resulting from IR (French et al., 2008). Superoxide dismutases (SOD) provide the primary cellular defenses against free radical accumulation by conversion of superoxide to hydrogen peroxide (H2O2) (Fridovich, 1997). Various isoforms of SOD have been identified, including copper/zinc SOD, mitochondrial manganese SOD (MnSOD or SOD2), and extracellular SOD (ecSOD) (Fridovich, 1997). Relative to the exercised heart, the mitochondrial isoform of superoxide dismutase (SOD2 or MnSOD) has been consistently shown to be upregulated following cardioprotective exercise, and to be essential for exercise induced cardioprotection (French et al., 2008). Importantly, the essentiality of SOD2 to the exercised phenotype has been clearly demonstrated through the application of antisense oligonucleotides directed against the endogenous antioxidant. Given the importance of SOD2 to long-term muscle function and health, these knockdown approaches from several groups indicate that a relatively modest (20%-50%) increase in SOD2 expression is protective against ventricular dysrhythmias and tissue infarct, but ventricular pump function (Yamashita et al., 1999; Hamilton et al., 2003; Lennon et al., 2004a). Extracellular SOD increases in response to exercise in the aortic endothelium, which is mediated by endothelial nitric oxide synthase (eNOS) (Fukai et al., 2000). The elevated ecSOD preserves nitric oxide (NO), and may contribute to exercise-derived cardioprotection (Fukai et al., 2000), as will be described in the next section. However, the role of extracellular ecSOD is unstudied in this context.
Early studies indicated that exercise training improves calcium handling during IR (Bowles et al., 1992). Although conflicts in the literature exist, some evidence indicates that exercise training improves mitochondrial tolerance to calcium overload, resulting in decreased mitochondrial permeability transition pore (mPTP) opening (Marcil et al., 2006; Starnes et al., 2007; Magalhaes et al., 2014). In addition, increased mitochondrial antioxidant concentration limits calcium overload that occurs during IR (Buja, 2005; Starnes et al., 2007). Indeed, exercise training increases SOD2, protecting against oxidative modification and calpain-mediated degradation of calcium handling proteins such as L-type calcium channels, phospholamban, and sarcoplasmic/endoplasmic reticulum calcium ATPase, which improves cardiac function and decreases tissue necrosis and apoptosis following IR (French et al., 2006; French et al., 2008). While these studies provide convincing evidence of the role of improved calcium handling as a key component of exercise-induced cardiac preconditioning, this area remains understudied relative to exercise-induced cardioprotection against ischemic insults.
Given that endogenous antioxidants are typically held to function as a network defense, conversion of H2O2 produced by SOD2 is likely to be just as important as the quenching of O2- directly. To this end, both the endogenous antioxidant catalase and the glutathione system, which function downstream of SOD to convert H2O2 to H2O and O2, have been extensively investigated. Catalase is inconsistently upregulated in exercised hearts, suggesting that components of the glutathione system may be an essential collaborator to SOD2 (Lennon et al., 2004b; Lennon et al., 2004a; French et al., 2008; Dao et al., 2011). Accordingly, myocardial glutathione reductase activity is enhanced following exercise and appears to serve as the essential second step for exercise induced cardioprotection for free radical generation of superoxide from the mitochondria during an IR challenge (Frasier et al., 2011). However, it appears that the other facets of the glutathione system do not need to up-regulate in parallel. Indeed, mixed results exist in terms of upregulation and requirement for protection by glutathione peroxidase, and it is proposed that improved ability to replenish glutathione by glutathione reductase (as opposed to increased glutathione peroxidase content) may be the mechanism responsible for glutathione system’s role in exercise cardioprotection (Frasier et al., 2011).
With regard to endogenous antioxidant function, it is important to note that just as IR pathology occurs through the parallel events of free radical production and calcium dyshomeostasis, exercise-induced protection mediates simultaneous protection through redundant mechanisms that offer tandem levels of protection. For instance, upregulation of antioxidant defense systems, and in particular SOD2, appear to benefit myocardial calcium handling following IR. It is currently unknown whether these observations are the result of directed antioxidant events in the cytosol or are due to the upstream quenching of free radical chain reactions. Nonetheless, it is clear that modulation of endogenous antioxidants, including SOD2, prevent oxidative modification of calcium handling proteins such as the sarcoplasmic endoplasmic calcium ATPase (SERCA2A) and other cellular indices of calcium dyshomeostasis (French et al., 2006; French et al., 2008).
Finally, while exogenous antioxidant compounds (e.g., high doses of vitamin E, vitamin C, glutathione, etc.) are often beneficial in limiting ischemic injury in the heart, they can mitigate the redox-sensitive adaptive response to exercise (Ristow et al., 2009). In this regard, an important investigation by the Powers lab combined exercise and an exogenous antioxidant cocktail (α-tocopherol, selenium, ascorbic acid, beta-carotene, CoQ10, N-acetyl cystine, and catechin) as a means of investigating the potentially additive effect against an ischemic insult. Findings revealed that exercise and antioxidants provided similar levels of independent cardioprotection, but that the combined dose of exercise and antioxidants did not potentiate the cardioprotective response (Hamilton et al., 2003). Further research is needed to resolve how long-term antioxidant dosing may alter exercise training outcomes, but as a preliminary conclusion it seems plausible that the exercised heart does not require the addition of antioxidant supplementation to bolster the cardioprotected phenotype.
Mitochondrial Proteins & Function
As described in the previous section, exercise induced cardiac preconditioning improves mitochondrial function during IR as a result of improved antioxidant status and calcium control (Bowles et al., 1992; French et al., 2006; Marcil et al., 2006; Starnes et al., 2007; French et al., 2008; Magalhaes et al., 2014). Subpopulations of subsarcolemmal and intermyofibrillar mitochondria exhibit distinct alterations following exercise, although more research is needed to fully understand these differences and functional relationship during IR (Kavazis et al., 2009). Both subpopulations from exercised hearts have decreased mPTP opening and decreased functional impairment during IR compared to the sedentary condition (Kavazis et al., 2008; Lee et al., 2012). In addition to elevated mitochondrial antioxidants previously discussed, such as SOD2, a decrease in the enzyme monoamine oxidase (MAO), which catalyzes amine oxidation reactions and the production of reactive oxygen species (ROS), appears to be another mechanism by which exercise protects the heart from IR (Kavazis et al., 2009; Deshwal et al., 2017). Collectively, exercise prior to IR protects against mitochondrial release of ROS, activation of apoptotic signaling molecules, and mitochondrial damage and functional impairment (Kavazis et al., 2008; Kavazis et al., 2009; Lee et al., 2012).
Nitric Oxide
Nitric oxide appears to be important for exercise-induced cardioprotection against IR induced arrhythmias and injury (Hajnal et al., 2005; Akita et al., 2007). The function and synthesis of NO has been reviewed extensively elsewhere (Suhr et al., 2013; Wu et al., 2018). The exercise mediated cardioprotective effects of NO include angiogenesis and ventricular remodeling after myocardial infarction with protective cellular signaling through opening of ATP-sensitive potassium channels (KATP) channels and a reduction in ROS (Wu et al., 2018). Nitric oxide also improves post-ischemic blood flow and contractile function (Pabla et al., 1995; Pabla et al., 1996). Nitric oxide production is enhanced following exercise training by increased activation and upregulation of eNOS (Fukai et al., 2000; Dao et al., 2011; Yang et al., 2014). Inducible nitric oxide synthase (iNOS) may also be involved in exercise mediated increases in NO, with some reports indicating that increases in eNOS following exercise training mediate elevated iNOS expression, which is required for exercise induced cardioprotection (Akita et al., 2007). However, not all studies have reported increases in iNOS (McGinnis et al., 2015). Additionally, as an inflammatory mediator, is likely not sustainable for a long period of time and some studies have indicated it is not essential for exercise induced cardiac preconditioning (Wilson et al., 2004; Quindry et al., 2010b; McGinnis et al., 2015). Activation of eNOS occurs via sympathetic nervous system activation and specifically β3-adrenergic receptors (Calvert et al., 2011). Elevated ROS and increased hydrogen peroxide also contribute to exercise mediated elevation in eNOS (Lauer et al., 2005; Akita et al., 2007). Although increases in antioxidant enzymes, such as genetic overexpression of catalase, result in diminished eNOS, catalase has not been consistently shown to increase following exercise training and eNOS remains increased following weeks of exercise training, indicating that any potential upregulation in antioxidant enzymes that might occur with exercise training are not inhibiting eNOS expression (Lauer et al., 2005; Dao et al., 2011; Yang et al., 2014). However, while exercise results in elevated eNOS in the myocardium and large arteries, this may not be the case in all tissues, as high catalase activity in venous tissue blocks exercise-induced increases in eNOS expression (Dao et al., 2011). Exercise-induced elevation of eNOS increases extracellular SOD, which then aids in maintaining NO levels by limiting superoxide mediated degradation of NO (Fukai et al., 2000).
Chaperone proteins and cardioprotection
An interesting facet of exercise and cardiac preconditioning is the lack of evidence for heat shock proteins (HSPs) as an essential mediator. Indeed, HPS expression is central to many forms of non-exercise preconditioning against ischemic insults, and exercise is well known to up-regulate HSPs, namely HSP72 (Demirel et al., 2003; Quindry et al., 2007). Nonetheless, experimental models have explored this topic using non-reductionist methods – performing treadmill exercise in a cold environment where the normal elevation in core temperatures is ~1.5-3 ºC. In these investigations, mitigation in HSP72 expression appeared to have no bearing on various observations of cardioprotection against ischemic insults (Hamilton et al., 2001; Demirel et al., 2003; Quindry et al., 2007). Moreover, HSP family proteins have also been investigated and observed to be completely dissociated from corresponding observations of exercise-induced cardioprotection against ischemic injury (Hamilton et al., 2001).
It should be acknowledged, however, that these experimental approaches do not preclude the potential importance of HSPs in exercise-induced cardioprotection. Indeed, redundant mechanisms of protection highlighted within may mask the role of HPSs in the cardioprotected phenotype. One intriguing possibility is that HSPs may be playing a tissue specific role within the heart, and the current methodological design are insufficient to delineate the importance of HSPs within ventricular tissue versus an independent role in perfusion vessels. In support, an interesting study by Melling et al. (2007) revealed through confocal microscopy that HSP overexpression in the exercised heart was localized to the vasculature. Accordingly, the possibility remains that HSPs could yet prove essential to exercise preconditioning in the heart and/or preserve vascular function during an ischemic challenge, although additional research is needed to confirm or refute this hypothesis. The reader is directed to a recent comprehensive review of the regulation of chaperone proteins as it pertains to cardiac preconditioning against IR (Penna et al., 2018).
Renin Angiotensin System
The renin angiotensin system (RAS) is comprised of two opposing arms: 1) the ‘classical’ arm, comprised of the angiotensin converting enzyme (ACE), and angiotensin I and angiotensin II receptors, and 2) ‘counter regulatory arm’ comprised of ACE2, the hepapeptide Ang-(1-7), and the Mas receptor (Ferrario et al., 1998; Donoghue et al., 2000; Nunes-Silva et al., 2017). Unlike the ‘classical’ arm, the ACE2/Mas receptor arm mediates anti-inflammatory, vasodilatory, and cardioprotective effects (Ferrario et al., 1998; Nunes-Silva et al., 2017). Exercise downregulates the classical RAS and upregulates the counter-regulatory RAS arm (Nunes-Silva et al., 2017; Magalhaes et al., 2020). More research is required to uncover the role of the dual arms of the RAS in exercise-induced cardiac preconditioning.
Channel function and improved metabolic control
Early investigations of exercise-induced cardioprotection using rat models of treadmill running and isolated heart preps revealed that calcium handling is significantly improved in exercised hearts (Bowles and Starnes, 1994). Indeed, parallel investigations of rats using isolated perfused working heart preparations from this group clearly indicate that improved calcium handling is inseparable from preservation of electron transport and tissue-level metabolism in exercised hearts (Bowles et al., 1992; Starnes and Bowles, 1995).
Roughly a decade later, it was discovered that KATP were also central to various forms of cardiac preconditioning of exercised hearts. Located on the sarcolemma (sarc KATP) and the inner mitochondrial membrane (mito KATP), the precise role of these putative mediators of cardioprotection is still debated (Light et al., 2001; Gross and Peart, 2003). Nonetheless, early findings in exercised hearts indicated that both sarc KATP and mito KATP are central to the exercised phenotype (Brown et al., 2005b; Johnson et al., 2006; Chicco et al., 2007). Interestingly, the only observations of sex-dependent differences in exercise-induced cardioprotection appear to relate to KATP channel sub-unit expression, where hearts from exercised females appear to over-express the Kir6.2 subunit to a different level than males (Johnson et al., 2006; Chicco et al., 2007).
While observations of variable KATP channel sub-unit expression in exercised hearts were also linked to comparable sex-dependent differences in infarct sparing, it appears that hearts from exercised males are nonetheless protected via KATP channel activation (Johnson et al., 2006; Chicco et al., 2007). To this end, and as mentioned earlier, the mechanisms of exercise-induced cardioprotection do not necessarily extend to all aspects of IR injury. Accordingly, an iterative series of experiments were performed where a common exercise regimen of treadmill running was applied against a variable duration of ischemic infarction as a means of evaluating the role of sarc KATP and mito KATP channels for preventing ventricular dysrhythmias and myocardial infarction. The shorter duration infarction study indicated that mito KATP channels were essential in preventing ventricular dysrhythmias, whereas the sarc KATP channel was essential for preventing post-infarct tissue necrosis and apoptotic processes (Quindry et al., 2010a; Quindry et al., 2012). These integrated findings are also important in that the ventricular dysrhythmia sparing effects observed following the shorter duration ischemic event were no longer present when the ischemic time was increased by 150% (Quindry et al., 2010a; Quindry et al., 2012). The interpretation of these findings is that while exercise robustly protects against the evolving insult of an IR challenge, the phenotype is providing a time-dependent tolerance that has finite limits.
Autocrine and paracrine mediated protection
Perhaps most important to the sub-field of exercise-induced cardioprotection is the notion that exercise preconditioning provides a pragmatic scientific lens through which a druggable counter therapy to mitigate IR injury could be discovered. While there are multiple approaches to condition an organ through exogenous means, receptor-mediated approaches are among the first to be considered. Accordingly, a number of groups interested in exercise-induced cardioprotection have asked the question of whether the redundant protection afforded to exercised hearts might include receptor mediated processes. Indeed, this rationale might indicate tissue-to-tissue cross talk confers cardioprotection through circulating factors.
Given the parallel roles of exogenous opioids as a temporary approach to pharmacologic cardioprotection (Gross and Gross, 2007), and the reputed fact that aerobic exercise sometimes produces an endorphin response, it was speculated that some common mechanisms may mediate exercise-induced cardioprotection (Schultz et al., 1997; Karck et al., 2001; Schultz and Gross, 2001; Maslov et al., 2009). Opioid peptides increase following an acute bout of exercise (Kraemer et al., 1985; Goldfarb et al., 1987), supporting the rationale that endogenous opioids could have a role in exercise-induced cardioprotection. Endogenous opioids include three major families: β-endorphins, dynorphins, and enkephalins, as well as at least three major opioid receptor subtypes, (µ, δ, κ), reviewed extensively elsewhere (van den Brink et al., 2003). Opioid peptides are released from various tissues, including the heart and skeletal muscle, with transcript expression of both opioid precursors and receptor subtypes increased in cardiac tissue following exercise (Denning et al., 2008; Dickson et al., 2008; Miller et al., 2015; Geng et al., 2017). Further, administration of a selective delta-opioid receptor agonist prior to exercise blunted the cardioprotective effect against tissue necrosis, suggesting the delta opioid receptor is involved in exercise induced cardioprotection (Miller et al., 2015). Some evidence also suggests that the kappa opioid receptor may also be involved in exercise induced cardioprotection, however not all studies have reported positive findings (Borges et al., 2014; Geng et al., 2017).
Although not definitive, it is most likely that opioid peptides (leu-enkephalin) released from the heart are mediating the cardioprotective effect (Schultz et al., 1997; Maslov et al., 2009; Miller et al., 2015). However, delta opioid receptors are located only on intrinsic cardiac adrenergic (ICA) cells within the myocardium, and the cardioprotective effect of delta opioid receptor activation appears to be mediated by the release of epinephrine and calcitonin gene related peptide from ICA cells acting upon the β2-adrenergic and calcitonin gene-related peptide (CGRP) receptors in cardiomyocytes (Huang et al., 2007; Huang et al., 2009). However, direct evidence that CGRP is released from ICA cells in response to exercise-stimulated opioid release is yet to be confirmed. Additionally, cardioprotection afforded by circulating factors following exercise resulted in loss of protection against tissue necrosis when a non-selective opioid antagonist was administered, indicating circulating opioid molecules may have a role in exercise induced cardioprotection (Michelsen et al., 2012). It may be that opioid-mediated cardioprotection involves both opioids directly produced in cardiac tissue as well as other tissues delivered via circulation and various opioid receptor subtypes, although these conclusions require confirmation (Michelsen et al., 2012; Miller et al., 2015).
In addition, more research is needed to identify the interaction between opioid peptides, receptors and downstream signaling processes involved in exercise induced cardioprotection. To this end, while it cannot be ruled out that endorphins responsible for the often described “runner’s high” may be cardioprotective, these factors are almost certainly not produced by the modest amounts of moderate intensity exercise observed to robustly protect against ischemic insults. This tentative conclusion is highly valuable to clinical and health-fitness narratives where patients and clients at significant risk for a heart attack can be assured that the best available scientific evidence indicates that one does not need to be a highly involved aerobic athlete to develop a cardioprotected heart.
Cytokines & Myokines
Extending the discussion of circulating factors and receptor-mediated cardioprotection, it is worth considering the role of cytokines/myokines and exercise-induced cardioprotection. By way of a brief review, inflammation is an important cellular response for survival and recovery. Yet for all the adaptive benefits of an acute inflammatory response of moderate intensity, it cannot be overlooked that dampening activation of inflammatory pathways in response to IR results in improved outcomes in terms of cell survival and function (Dawn et al., 2004). Central to this line of research is the molecule, interleukin-6 (IL-6). While IL-6 is traditionally held to be a potent cytokine, promoting inflammatory responses with significant collateral damage to healthy tissues, more recent evidence indicates that IL-6 may also have anti-inflammatory roles. While the full understanding of a dual functioning IL-6 is still being resolved, it appears that its role is contextual to the cytokine milieu. Thus, following a pathological stressor such as IR, the cytokine profile, which includes tumor necrosis factor alpha (TNFα), results in IL-6 serving a deleterious role. However, following an acute bout of exercise, IL-6 is released in vesicles from contracting skeletal muscle, serving as an anti-inflammatory myokine, producing beneficial adaptative responses (Pedersen, 2009; Karstoft and Pedersen, 2016).
While not extensively examined in the context of exercise-induced cardioprotection, one investigation provides preliminary insights. Using a whole-body IL-6 knockout mouse model of exercise and surgical LAD ligation, it was observed that IL-6 may be an important mediator of exercise preconditioning against both ventricular dysrhythmias and tissue death. Interestingly, the short-term exercise model resulted in over-expression of both soluble (circulating) and insoluble IL-6 receptors in the myocardium (McGinnis et al., 2015). This finding was interpreted to suggest that the benefits linking IL-6 function to exercise preconditioning may also confer cytoprotection to other tissues throughout the body through co-activation of IL-6 family receptor sub-types.
Exosomes
Recent studies suggest that exosomes, which transport various signaling molecules such as miRNAs, proteins, and lipids, may confer cardioprotective signals (Record et al., 2014; Vicencio et al., 2015; Hou et al., 2019). Some evidence suggests that exercise may confer protection against IR by increasing the circulating exosome concentration (Whitham et al., 2018). Indeed, exosomes isolated from plasma following exercise decreased infarct size and improved cardiac function following an experimental model of IR, although no differences in exosome number were found following exercise intervention (Hou et al., 2019). Specifically, exosomal miR-342-5p has been found to contribute to exercise derived cardioprotection by inhibiting apoptosis and promoting cell survival (Hou et al., 2019). Although most cell types secrete exosomes, it seems that vascular endothelial cells may be primarily responsible for increases in exosome miR-342-5p, which evidence indicates that flow-induced shear stress that occurs during exercise, acts as the signal (Hou et al., 2019). Additionally, it appears that cardiomyocytes have the ability to take up vascular endothelium derived exosomes, although others have proposed ligand-receptor mediated signaling (Vicencio et al., 2015; Hou et al., 2019). Exosome mediated cardioprotection has been demonstrated to involve HSP70 in models of non-stressed exosome (Vicencio et al., 2015). Cardiac upregulation of HSP70 has been found to not be essential for exercise induced cardioprotection (Hamilton et al., 2001; Quindry et al., 2007). It could be that protective pathways are activated in cardiomyocytes by exosomal HSP70 (Vicencio et al., 2015), opposed to upregulation within the cardiomyocyte. However, whether exercise cardioprotection involves exosomal associated HSP70 is currently unknown. Exosomes appear to be involved in various forms of cardiac preconditioning, such as remote ischemic preconditioning (Giricz et al., 2014; Vicencio et al., 2015). Future work in this exciting field to uncover mechanisms of exosome-derived cardioprotection are warranted.
Evidence from human studies
Evidence supporting exercise induced preconditioning in humans has been recently reviewed elsewhere (Thijssen et al., 2018) and will be briefly summarized here. Retrospective studies indicate improved survival rates for individuals with greater physical activity levels prior to an acute myocardial infarction and percutaneous transluminal coronary angioplasty (Abete et al., 2001; Rengo et al., 2007). Additionally, physical activity prior to coronary artery bypass graft improved outcomes by increasing survival rate (Rengo et al., 2010).
Human studies suggest a protective effect of previous exercise on angina and ST segment depression during a subsequent exercise session, referred to as ‘warm-up angina’. It appears an initial stress test can decreased angina, later onset of ST segment depression, and decreased peak ST segment depression during a second stress test, lending support for the rationale that a single exercise session is protective against future myocardial ischemia in humans with angina (Lalonde et al., 2015). In contrast, the relatively moderate dose of exercise used in the majority of animal-based studies (without coronary artery disease) does not require the myocardium to become ischemic. However, mild to moderate exercise below the ischemic threshold was found to increase exercise duration and postpone angina onset, although this was related to decreased heart rate and blood pressure reactivity, so when compared to rate pressure product the angina onset was unchanged by moderate exercise, and only improved with relatively intense ischemia in the first exercise session (Bogaty et al., 2003). Still, existing evidence suggests that exercise preconditioning occurs in humans.
Conclusions and future directions
The field of exercise preconditioning continues to expand with important implications for both scientific exploration and clinical application. Due to the sustainable nature, effectiveness across the sexes and age groups, and evidence of protection in human clinical populations, more research and efforts for clinical application are needed. Although currently supported by human studies, additional work to clarify the specific exercise requirements and potential of exercise as a preconditioning stimulus prior to planned cardiac surgical procedures is warranted. Additionally, many preliminary findings detailed herein serve as a scientific rationale for the successful reverse engineering of cardioprotective counter therapies against IR injury. Perhaps just as important, exercise programming should be included in tandem with pharmacological prescriptions to treat and prevent cardiovascular and metabolic diseases. To this end, the reader is directed to the American College of Sports Medicine’s Exercise is Medicine platform (https://exerciseismedicine.org). Exercise is Medicine encompasses a wealth of refereed research detailing the cooperative interface between an exercise lifestyle and modern personalize medicine. Indeed, the tenets of Exercise is Medicine extend well beyond the current topic of exercise-induced cardiac preconditioning. In this regard however, the sub-field of exercise preconditioning is among the more scientifically progressive research lines, and it is tempting to speculate that many of the cytoprotective mechanisms already identified in the exercised heart are also responsible for the preconditioning effects descriptively observed in other organ systems throughout the body.
Conflict of Interest Statement:
The Authors declare that they have no conflicts of interest.
References
Lindsey E. Miller1
1Department of Nutrition and Exercise Physiology, Elson S. Floyd College of Medicine, Washington State University Health Science Center Spokane, Spokane WA.
John C. Quindry2,3
2Integrative Physiology and Athletic Training, University of Montana, Missoula, MT. 3International Heart Institute, St. Patrick's Hospital, Missoula, MT.
Corresponding author:
Lindsey E. Miller
Email: Lindsey.e.miller@wsu.edu
In a new window | Download PPT
Figure 1: Ischemia reperfusion injury. Ischemia-reperfusion results in arrhythmia, decreased cardiac function, and cell death. Elevated free radical production, calcium overload, and decreased mitochondrial function are central to ischemia-reperfusion injury. Figure created by modification of an image created by Patrick J. Lynch, medical illustrator; C. Carl Jaffe, MD, cardiologist. https://creativecommons.org/licenses/by/2.5/
In a new window | Download PPT
Figure 2: Mechanisms of exercise induced cardioprotection against ischemia-reperfusion injury. Exercise prior to ischemia-reperfusion decreases arrhythmia, improves cardiac function, and decreases cell death. Short-term exercise induces protective biochemical alterations within the cardiomyocyte, which are summarized here. SOD2, mitochondrial isoform of superoxide dismutase; GR, glutathione reductase; MAO, monoamine oxidase; MPTP, mitochondrial permeability transition pore; NO, nitric oxide; eNOS, endothelial nitric oxide synthase; SERCA2A, sarcoplasmic endoplasmic calcium ATPase; KATP, ATP-sensitive potassium channels; IL-6, interleukin-6, sIL-6R, soluble IL-6 receptor; ICA, intrinsic cardiac adrenergic; CGRP, reticulum calcitonin gene related peptide.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11129 | 30 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA