Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
[Special Issue] Therapeutic potential of quercetin in cardiovascular and neuromuscular disorders
Time:2020-07-03
Number:14505
Author Affiliations

Conditioning Medicine 2020. 3(3):117-134.
Abstract
Chronic conditions including cardiovascular and neuromuscular disorders represent major causes of morbidity and mortality globally. Thus, effective therapeutic interventions are necessary to extend longevity and improve quality of life. One such intervention includes dietary enrichment with flavonol compounds. Flavonols are a large family of naturally occurring phytochemicals concentrated in many edible plants. One of the most abundant flavonols is quercetin, which includes its naturally occurring conjugates (i.e. isoquercetin, rutin). Quercetin has demonstrated antioxidant, anticarcinogenic, and anti-inflammatory properties and can activate molecular mediators of metabolic adaptation in skeletal and cardiac muscle. Because etiologies of cardiac and skeletal muscle disorders are often multi-faceted, a single therapeutic agent that targets multiple pathological processes is of great practical importance. Moreover, treatment efficacy is predicated on the notion that such an intervention be indefinitely sustainable and safe during chronic use while also remaining potent in aged individuals. In this review, we provide emergent evidence to suggest that supplemental use of quercetin may contribute to mitigation of a wide range of cardiovascular and neuromuscular conditions through various physiological mechanisms. In particular, the molecular underpinnings of therapeutic potential of quercetin in disease will be emphasized and potential implications for clinical therapy will be discussed.
Keywords: flavonol, muscular dystrophy, ischemia-reperfusion injury, heart disease, skeletal muscle
Abstract
Chronic conditions including cardiovascular and neuromuscular disorders represent major causes of morbidity and mortality globally. Thus, effective therapeutic interventions are necessary to extend longevity and improve quality of life. One such intervention includes dietary enrichment with flavonol compounds. Flavonols are a large family of naturally occurring phytochemicals concentrated in many edible plants. One of the most abundant flavonols is quercetin, which includes its naturally occurring conjugates (i.e. isoquercetin, rutin). Quercetin has demonstrated antioxidant, anticarcinogenic, and anti-inflammatory properties and can activate molecular mediators of metabolic adaptation in skeletal and cardiac muscle. Because etiologies of cardiac and skeletal muscle disorders are often multi-faceted, a single therapeutic agent that targets multiple pathological processes is of great practical importance. Moreover, treatment efficacy is predicated on the notion that such an intervention be indefinitely sustainable and safe during chronic use while also remaining potent in aged individuals. In this review, we provide emergent evidence to suggest that supplemental use of quercetin may contribute to mitigation of a wide range of cardiovascular and neuromuscular conditions through various physiological mechanisms. In particular, the molecular underpinnings of therapeutic potential of quercetin in disease will be emphasized and potential implications for clinical therapy will be discussed.
Keywords: flavonol, muscular dystrophy, ischemia-reperfusion injury, heart disease, skeletal muscle
1. Introduction
Chronic conditions such as cardiovascular (CVD) and neuromuscular (NMD) disorders represent a wide range of heterogenous pathologies leading to increased morbidity and pre-mature mortality globally (Johnson et al., 2014). In 2017, 12.1% of all adults in the United States were given a new CVD diagnosis while NMD prevalence has been reported to occur as high as 10 per 100,000/year (Deenen et al., 2015; Blackwell and Villarroel, 2018). While myopathy and cardiopathology are commonly mediated by underlying genetic and hereditary factors, pathologies may be attenuated or rescued by pharmaceutical or nutritional interventions and lifestyle modifications. In addition to conventional medicines, more than 250,000 natural supplements have been purported to treat or prevent chronic diseases, many without scientific confirmation. Nonetheless, sales within the global dietary supplement market are estimated to reach 133.1 billion USD by 2027 (Helal et al., 2019). However, there is growing concern and criticism over regulation and efficacy of many supplements currently on the market. Thus, more comprehensive reviews of literature regarding supplementation with individual supplements are warranted in order to determine therapeutic potential.
Derived from food, nutraceuticals represent a large class of compounds that may enhance health and improve both physical and mental function (Helal et al., 2019). While there is no consensus on a clear definition, nutraceuticals have been described as food or parts of food that provide medicinal or health benefits, including the prevention and/or treatment of disease (DeFelice, 1995). Dietary nutraceutical enrichment has been suggested to alleviate and/or attenuate inflammatory, metabolic, cancer, and cardiac conditions, as well as offset some age-associated changes (Helal et al., 2019). While nutraceuticals are regulated in Europe by the European Food Safety Authority (EFSA), most other parts of the world have limited regulations on nutritional supplements. For example, the Food and Drug Administration (FDA) in the United States only requires: “This statement has not been evaluated by the FDA. This product is not intended to prevent, cure or treat any disease” (Frankos et al., 2010). Overall, many nutraceuticals lack scientific support by blinded, randomized, placebo-control trials. However, polyphenols, a class of phytochemicals, have been amassing scientific backing as potent nutraceuticals with hundreds of controlled studies reporting therapeutic benefit over the past decade (Watson et al., 2018).
Polyphenols are naturally occurring molecules characterized by the presence of multiple phenol groups around their core structure (Haslam and Cai, 1994). Occurring almost exclusively in plants, polyphenols represent one of the largest dietary sources of antioxidants (Scalbert et al., 2005). Hundreds of polyphenols have been characterized and are often concentrated in rinds, skins, and leaves of edible plants. Within the flavonoid form, classes include flavones, isoflavones, flavanones, anthocyanins, and flavonols (Scalbert et al., 2005). While intake in humans varies by diet and geographical location, flavanol consumption ranges from 6-60 mg/day (Hollman and Arts, 2000).
Quercetin (3,3’,4’,5,7-pentahydroxyflavone) represents one of the most abundant flavanols consumed within modern diets. Multiple investigations report that quercetin consumption values average ~20 mg/day through normal food choices (Nishimuro et al., 2015; Yao et al., 2019). Thus, scientific interest in quercetin as a nutraceutical has increased over the last 10-15 years. Like other flavonols, quercetin has potent antioxidant effects and improves endogenous antioxidant enzyme function (Chang et al., 1993; Ballmann et al., 2017). Furthermore, improvements in metabolic function and activation of oxidative transcriptional mediators have been reported by multiple groups (Eid et al., 2010; Ballmann et al., 2016). Anti-inflammatory and carcinogenic effects of quercetin have also been documented (Lesjak et al., 2018). Altogether, quercetin is one of the most widely studied flavonols and its use as a nutraceutical is commonly supported in animal and in many human studies. Given that cardiovascular and neuromuscular pathologies are multi-faceted, the pleiotropic nature of quercetin’s effects make it an appealing candidate to combat contributors of disease. To date, dietary quercetin enrichment has been shown to attenuate and/or rescue multiple pathologies and diseased states (Lakhanpal and Rai, 2007). In the following review, we provide both past and current physiological evidence for the possibility of quercetin therapy to attenuate underlying causes of CVD and NMDs. Underlying biochemical and molecular mechanisms responsible for medicinal actions are emphasized and future directions and clinical implications are discussed.
Quercetin
Chemical structure and class
Quercetin is a naturally occurring polyphenolic flavonoid in a sub-class of flavonoids called flavonols (Lakhanpal and Rai, 2007). When chemically isolated, quercetin is a dark-yellow, crystalline solid with a bitter taste. Solubility in water is poor but it is soluble in alcohol and alkaline solutions (Chebil et al., 2007; Razmara et al., 2010). Like other flavonols, quercetin has a double benzene core connected by a heterocyclic pyrone ring. Additionally, quercetin has 5 hydroxyl (OH) groups, which may be responsible for much of its antioxidant properties (Husain et al., 1987; Chen et al., 1996). These groups are located at the 3, 5, 7, 3', and 4' positions (Figure 1). Quercetin is not produced endogenously by animals but is widely synthesized by many plant species with concentrations being highest in rinds and barks (Lakhanpal and Rai, 2007). Derived from phenylalanine, quercetin is created by a series of steps in the phenylpropanoid pathway in combination with malonyl-CoA and eventually forms dihydroquercetin, which is turned into quercetin via flavonol synthase (Winkel-Shirley, 2001). In pure form, quercetin is an aglycone lacking any attached saccharides (Kelly, 2011). However, quercetin, as well as other flavonols, are often found in glycone forms (Herrmann, 1976). This is of particular importance as bioavailability may be dependent on quercetin’s glycosylation state (Hollman et al., 1997; Morand et al., 2000). Examples of common naturally occurring glycone forms of quercetin include quercetin-3-glucuronide, quercetin-3-rutinoside, and quercetin-3-L-rhamnoside (Kovalev and Litvinenko, 1965; Bloms et al., 2016)
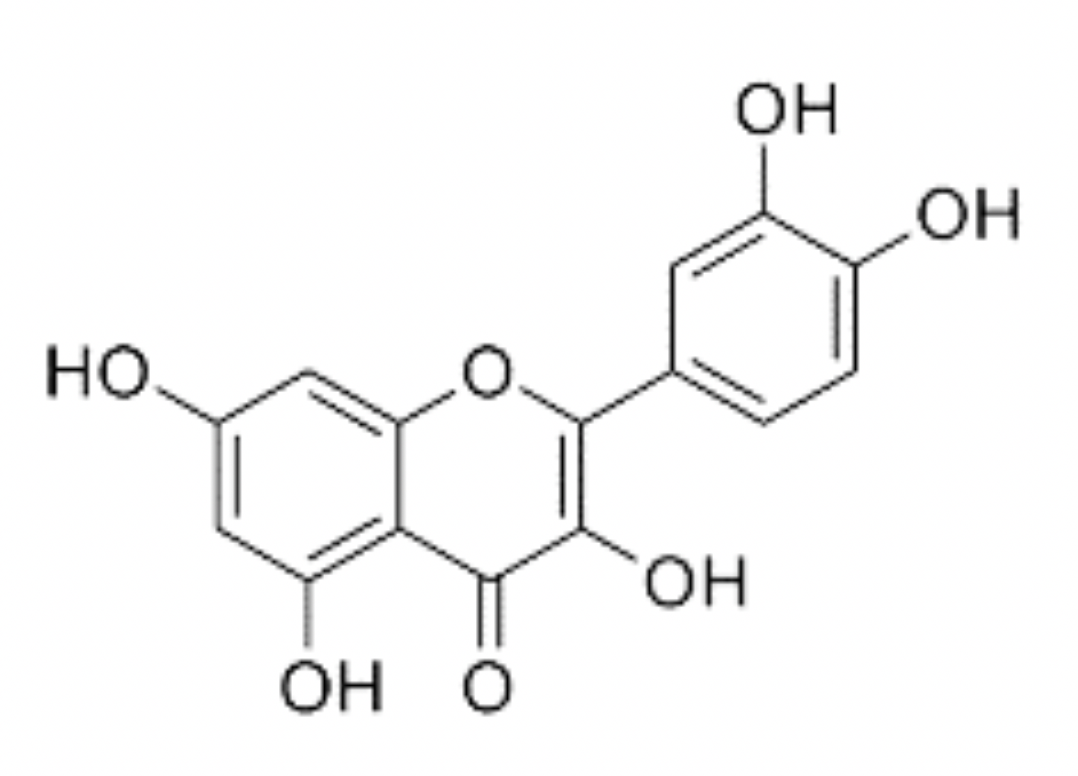
In a new window | Download PPT
Figure 1: Chemical structure of quercetin. IUPAC 2- (3,4-dihydroxyphenyl)-3,5,7 trihydroxychromen-4-one. Molecular weight: 302.23 g/mol.
Dietary sources
Quercetin is abundant in many different types of edible plants. Quercetin and glycone conjugates are found in onions, teas, herbs (i.e. oregano, cilantro), apples, capers, and a variety of berries (Bhagwat et al., 2011). Examples of food sources with moderate to high quercetin levels and concentrations are presented in (Table 1). While quercetin aglycone is present in many edible plants, the majority of ingested quercetin exists in glycosylated forms (Bilyk et al., 1984; Hollman et al., 1995). Quercetin form and concentration from dietary sources are dependent on multiple factors including the part of the plant eaten and how it is prepared. For example, quercetin content in varieties of onion (Allium cepa L.) is highest in outer dry layers and decreases with each inner layer (Patil and Pike, 1995). Furthermore, onion (Allium cepa L.) quercetin conjugate content increases 7-25% by sautéing/baking and decreases by 18% upon boiling cooking preparations (Lombard et al., 2005). Thus, these aspects should be considered when determining dietary flavonol consumption. Average daily consumption of quercetin ranges from ~16- 24 mg·day-1 (de Vrie et al., 1997; Nishimuro et al., 2015) though may vary seasonally (Nishimuro et al., 2015), by diet (i.e. western, Mediterranean, etc.), and geographic location (de Vrie et al., 1997). Overall, quercetin is present in physiologically appreciable amounts in most diets and is one of the most abundant flavonoids in food sources. Importantly, quercetin safety is widely accepted and doses as high as 1,900 mg/kg/day for 14 months resulted in no clinical sign of toxicity in male and female F344/N rats (Dunnick and Hailey, 1992). Furthermore, quercetin has achieved GRAS status by the FDA (GRAS Notice No. GRN 000341).
Table 1. Common foods containing quercetin and quercetin conjugates. All values are reported based on the USDA database for the flavonoid content of selected foods (Bhagwat et al., 2011).

Bioavailability and Metabolism
Early investigations showed extremely poor bioavailability of quercetin after a single oral dose, with absorption rates of less than 1% (Gugler et al., 1975). However, quercetin bioavailability has high inter-subject variability and is dependent on numerous factors (Kaushik et al., 2012; Kaşıkcı and Bağdatlıoğlu, 2016). First, aglycone quercetin is relatively insoluble in an aqueous solution, limiting absorption if quercetin is not solubilized before or during administration. Indeed, absolute bioavailability of quercetin aglycone is enhanced to ~45% when solubilized in solvents such as ethanol (Walle et al., 2001). Given that quercetin aglycone is lipophilic, high dietary fat intake with supplementation can also increase quercetin plasma concentrations by up to 32% as compared to low fat intake (Guo et al., 2013). Recent evidence indicates that delivery systems may differentially influence bioavailability. Self-double-emulsifying drug delivery systems (SDEDDS) increase the efficacy of absolute bioavailability of quercetin and extend-release (Wang et al., 2017). Furthermore, nanoparticle and nanocarrier delivery systems also increase the efficacy of absorption (Penalva et al., 2017; Peñalva et al., 2019). Limited evidence is available as to whether nanocarriers improve long-term outcomes in CVD and NMD but newer investigations suggest possible benefits and targeted delivery to specific disease sites (Fuior and Calin, 2020).
Arguably the most important factor in bioavailability during quercetin supplementation is the form ingested. Whereas the aglycone form of quercetin is typically poorly absorbed on its own, glycone forms appear to be more readily absorbed. As previously mentioned, quercetin glycosides are the primary form existing in plants and conjugated forms have higher bioavailability (Kelly, 2011; Lee and Mitchell, 2012). Quercetin-3-glucoside, one of the most commonly occurring quercetin glycone forms, yields ~55% higher plasma quercetin concentrations versus quercetin aglycone supplementation (Cermak et al., 2003). Quercetin-3-O-rutinoside also has higher absolute bioavailability compared to quercetin aglycone and results in a more sustained absorption (Reinboth et al., 2010). This outcome likely contributes to previous evidence showing greater quercetin absorption from edible plants versus some supplemental forms (Hollman et al., 1995; Hollman et al., 1997; Guo and Bruno, 2015). Indeed, naturally glycosylated quercetin conjugates from red onions are absorbed ~15 fold more than aglycone dietary supplemental forms (Shi and Williamson, 2015)
Quercetin aglycone is primarily absorbed in the stomach while glycoside forms are not absorbed by gastric tissue (Crespy et al., 2002). Both aglycone and glycone forms are absorbed in the small intestine (Crespy et al., 1999; Carbonaro and Grant, 2005). Solubilized glycoside forms appear to be deglycosylated to aglycone forms and then primarily diffuse passively or are secondarily transported across the intestinal epithelium (Crespy et al., 1999; Chabane et al., 2009; Guo and Bruno, 2015). Bioavailability may be higher with glycone forms due to glycoside-dependent transport via sodium-dependent glucose transporters (SGLT1) (Walgren et al., 2000). Once absorbed, quercetin is modified (i.e. methylation, sulfonation) and the primary derivatives in plasma are quercetin-3-glucuronide, 3’-methyl-quercetin-3-glucuronide and quercetin-3’-sulfate (Day et al., 2001). These derivatives are taken up by various cells and tissues including fibroblasts, gastrointestinal, skeletal muscle, and cardiac tissues where they then exert cellular effects (Spencer et al., 2003; Graf et al., 2006; Eid et al., 2010; Daubney et al., 2015). Cellular action of quercetin metabolites appears to be dependent on how quercetin has been modified, as some forms (i.e. methylated) may be more readily taken up by cells than others (Spencer et al., 2003). Once taken up, quercetin-induced cellular activation of metabolic secondary messengers occur including 5’ adenosine monophosphate-activate protein kinase (AMPK), sirtuin 1(SIRT1)/peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α), and AKT, which will be the topic of the subsequent sections. Peak quercetin plasma concentrations after a single quercetin-rich meal containing both aglycone and glycone forms appear at approximately 3 hours after ingestion while elimination half-life is approximately 17 hours (Hollman et al., 1997). Ingestion of pure quercetin glycosides appears to cause faster peak plasma levels (approximately 30 minutes) while elimination half-life remains similar to quercetin rich meals (approximately 18 hours) (Olthof et al., 2000). Unabsorbed quercetin is excreted in the feces and quercetin metabolites are almost completely eliminated in bile and urine with 48 hours (Ueno et al., 1983). While not central to the current review, excretion of unabsorbed quercetin may also benefit health through the prevention of colon cancer (Darband et al., 2018).
Biochemistry and Molecular Targets
The apparent potency of quercetin is predicated on bioactivity with several molecular targets, which are critical in the prevention of disease processes. Indeed, consumption of quercetin can alter cellular biochemistry such that bioenergetic control, inflammation, and redox reactions are favorably altered. In this regard, specific attention will now be focused on activation of AMPK, SIRT1/PGC-1α, activation of matrix metalloproteinases (MMPs), and attenuation of oxidative stress.
5' adenosine monophosphate-activated protein kinase (AMPK)
AMPK is an enzymatic protein complex that is highly conserved among eukaryotes. While AMPK has a multitude of functions, activation is dependent on elevated adenosine monophosphate:adenosine triphosphate (AMP:ATP) ratio thus being important in maintaining energy homeostasis (Hardie et al., 2012). Furthermore, increased intracellular calcium and reactive oxygen species (ROS) influence AMPK activity (Emerling et al., 2009; Park et al., 2013). Through direct or indirect mechanisms, AMPK regulates mitochondrial biogenesis, cellular glucose transport, mitophagy, and antioxidant enzyme production in the heart and skeletal muscle (Rabinovitch et al., 2017; Herzig and Shaw, 2018; Lin and Hardie, 2018). Quercetin has been repeatedly shown to induce AMPK activation (Ahn et al., 2008; Shen et al., 2012). While mechanisms by which quercetin mediates AMPK activation are not fully understood, quercetin has been shown to increase the AMP:ATP ratio in isolated cells (Dong et al., 2014a), which leads to increased AMPK activity. This is likely since quercetin, and other polyphenolic molecules, have been shown to partially inhibit mitochondrial function acutely and increase mitochondrial uncoupling (Nabavi et al., 2015; Choi et al., 2018). Furthermore, quercetin-initiated activation of AMPK may be due to inhibition of calcium reuptake into the sarcoplasmic reticulum as demonstrated by quercetin treatment in skinned muscle fibers (Shoshan et al., 1980). Once activated by quercetin, AMPK has been shown to increase autophagy and decrease cellular injury (Chen et al., 2014). Evidence has revealed quercetin-mediated AMPK activation increases glucose transporter (GLUT4) translocation and glycogen synthesis in liver and skeletal muscle (Eid et al., 2015). AMPK-dependent increases in mitochondrial membrane potential, ATP content, and biogenesis have also been reported as a consequence of quercetin administration (Qiu et al., 2018). Many of these actions by AMPK are a consequence of downstream interaction with SIRT1 and PGC-1α molecules (Figure 2).
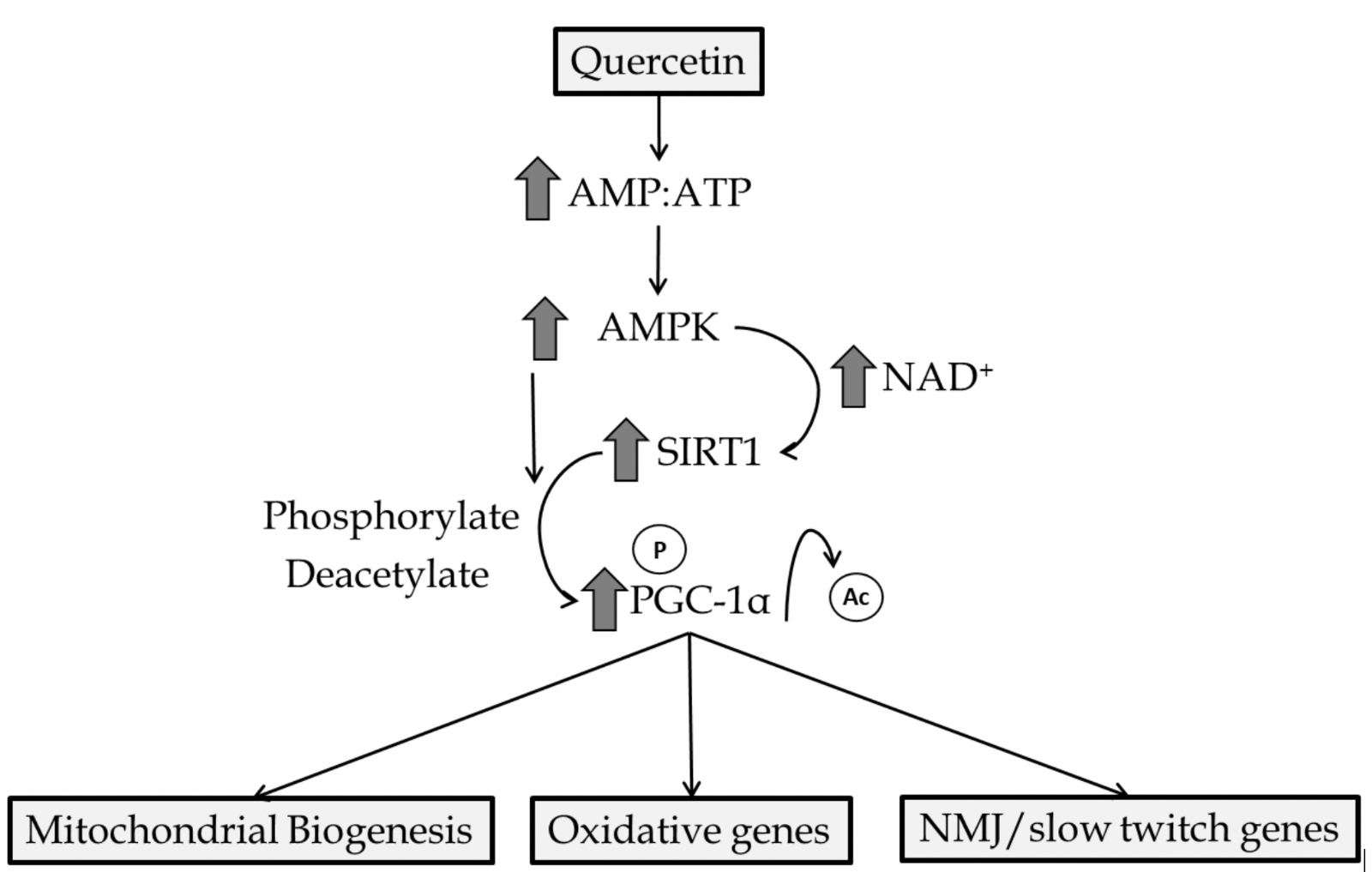
In a new window | Download PPT
Figure 2: Primary quercetin mediated activation of AMPK/SIRT1/PGC-1α. Creation of an acute energy supply demand mismatch activates AMPK and increases intracellular NAD+ concentrations. AMPK leads to phosphorylation and/or the latter results in SIRT1 activation leading to the deacetylation of PGC-1α. Genes involved in mitochondrial biogenesis, oxidative metabolism/antioxidant enzyme production, and slow-twitch genes are upregulated by PGC-1α interaction with nuclear receptors.
SIRT1 and PGC-1α
SIRT1 is an NAD-dependent deacetylase that is highly conserved and expressed across most biological species (Sinclair and Guarente, 2006). SIRT1 is highly sensitive to changes in nicotinamide adenine dinucleotide (NAD+) levels and plays a critical role in cellular control and maintenance of energy homeostasis. Whether direct or indirect, SIRT1 regulates oxidative/fat metabolism, mitochondrial function, antioxidant capacity, and overall life-span of organisms (Radak et al., 2020). In part, this is achieved through interaction and activation of PGC-1α via SIRT1-mediated deacetylation of PGC-1α to initiate transcription of genes involved in oxidative metabolism, mitochondrial biogenesis, neuromuscular junction (NMJ), and fast to slow fiber-type shift (Price et al., 2012). PGC-1α can also be activated through phosphorylation by AMPK among other factors (i.e. calcineurin, p38) (Rowe et al., 2010). PGC-1α, originally characterized in brown adipose tissue (Puigserver et al., 1998), is a transcriptional co-activator, which largely binds to nuclear receptors (i.e. estrogen-related receptors, peroxisome proliferator-activated receptors, nuclear respiratory factors) enhancing transcriptional activity leading to augmentation of fat oxidation, angiogenesis, mitochondrial fission/fusion, and mitochondrial biogenesis (Rowe et al., 2010). Neuromuscular junction genes (i.e. utrophin, MuSK, RAPSN) have been shown to be upregulated with PGC-1α overexpression (Handschin et al., 2007; Selsby et al., 2012). Activation and increased expression of PGC-1α has been suggested to alleviate multiple CVDs and NMDs including diabetic cardiomyopathy (Sulaiman et al., 2010), dystrophic cardiac dysfunction (Ballmann et al., 2016; Ballmann et al., 2017), doxorubicin cardiotoxicity (Liu et al., 2019), muscular dystrophy (Handschin et al., 2007; Selsby et al., 2012), and disuse atrophy (Wang and Pessin, 2013). Alternatively, constitutive overexpression of SIRT1/PGC-1α has been linked to several negative outcomes in cardiac and skeletal muscle (Miura et al., 2006; Kawashima et al., 2011). In part, this may be due to increased muscular fat accumulation as well as uncontrolled mitochondrial biogenesis leading to eventual loss of sarcomere structure (Lehman et al., 2000). To this end, polyphenols with a similar biochemical profile, namely resveratrol and quercetin, have been repeatedly shown to safely activate the SIRT1/PGC-1α axis (Davis et al., 2009; Price et al., 2012; Ballmann et al., 2016). Given that quercetin and resveratrol increase cellular the AMP:ATP ratio (Price et al., 2012; Dong et al., 2014b), this common observation is likely due to the aforementioned interaction with AMPK and increases in intracellular NAD+ concentrations. Indeed, AMPK and SIRT1 activation are closely dependent on one another and are pivotal in adaptive responses to polyphenol administration (Price et al., 2012). Quercetin-induced PGC-1α expression (and likely activity) has been shown to be increased in a variety of tissues including the brain, heart, and skeletal muscle following quercetin administration (Davis et al., 2009; Ruiz et al., 2015). Quercetin administration increases antioxidant enzyme production, improve mitochondrial function, and decrease inflammatory responses (Henagan et al., 2014; Ballmann et al., 2017). Increased cytoskeletal and NMJ proteins (i.e. α-sarcoglycan, utrophin) have also be reported with long-term quercetin supplementation with concomitant increases in PGC-1α protein expression (Ballmann et al., 2017). Thus, it appears that much of quercetin’s beneficial metabolic effects are exerted through activation of the SIRT1/PGC-1α axis and have important implications for using quercetin as a nutraceutical in a variety of cardiovascular and neuromuscular pathologies.
MMPs
MMPs are a family of calcium-dependent endopeptidases that are important in the remodeling and degradation of extracellular matrix (ECM) proteins in various tissue types (Verma and Hansch, 2007). There are 23 known variants of MPPs (Nagase et al., 2006), including MMP-1, MMP-2, and MMP-9. MMP-1 is a collagenase that degrades interstitial collagen including type I, II, and III collagen (Verma and Hansch, 2007). As a family of proteins, MMPs have been postulated to play a role in cellular migration/adhesion, angiogenesis, and progression of atherosclerosis (Lee et al., 1996; Sang, 1998; Blackburn et al., 2007). MMP-1-mediated angiogenesis may be due to the creation of extracellular space allowing vessel branching (Sang, 1998). Work in in vitro and in vivo models has shown quercetin administration can differentially influence expression of MMP-1 (Song et al., 2001; Cho et al., 2010). Fibrotic tissue proliferation and development are blunted by quercetin via increased MMP-1 activity (Cho et al., 2010). Quercetin inhibits oxidized-low density lipoprotein-induced MMP-1 expression in cell models of atherosclerotic plaque formation indicating possible attenuation of plaque destabilization (Song et al., 2001). Thus, quercetin may mediate fibrotic and atherosclerotic processes through the modulation of MMP-1. MMP-2 and MMP-9 are gelatinases for which primary substrates are gelatin and collagen type IV (Verma and Hansch, 2007). In skeletal muscle, both MMP-2 and MMP-9 are upregulated after injury and are involved in regeneration in a fiber-type specific manner (Zimowska et al., 2003). However, blockade of MMP-2 and MMP-9 attenuates development of pathological fibrosis in skeletal muscle suggesting a role for MMP exacerbation of skeletal muscle pathology (Gosselin et al., 2007; Morris et al., 2010). Both MMP-2 and MMP-9 have been implicated in pathologic cardiac remodeling and fibrosis (Reinhardt et al., 2002; Van Linthout et al., 2008). Sarcomere degradation in dilated cardiomyopathy is associated with elevated MMP-2 and MMP-9 activity suggesting MMPs may be key mediators in contractile loss during cardiac pathology (Rouet-Benzineb et al., 1999). Quercetin has been repeatedly shown to decrease MMP-2 and MMP-9 activation and expression (Vijayababu et al., 2006; Ballmann et al., 2016; Ballmann et al., 2017). Inhibition of MMP-2 and MMP-9 via quercetin are associated with decreased fibrotic markers (Yoon et al., 2012). Doxorubicin-induced cardiac MMP-2 activation is blunted with quercetin supplementation and results in improved ischemic tolerance (Barteková et al., 2015). Furthermore, quercetin-mediated decreases in MMP-9 activity have been shown to attenuate pathological cardiac remodeling and preserve cardiac function in dystrophic cardiac pathology (Ballmann et al., 2016; Ballmann et al., 2017). Taken together, quercetin appears to modulate pathological remodeling of both skeletal and cardiac muscle at least in part through interactions with MMPs.
Antioxidant enzymes and properties
Reactive oxygen species (ROS) and oxidative stress have been shown to exacerbate both neuromuscular and cardiovascular pathology (Wallace, 2000). ROS are highly reactive free radical species that can cause damage to various cellular constituents including DNA, proteins, and lipids (Halliwell, 1991). Mitochondria are major sources of ROS through partial reduction of oxygen, leading to the formation of superoxide anions (O2-), which are dismutated to hydrogen peroxide (H2O2) (Murphy, 2009). Cytosolic enzymes (i.e. monoamine oxidase, xanthine oxidase) can also produce free radicals (Halliwell, 1991). Neutralization of excessive free radical formation and prevention of oxidative damage is essential in maintaining optimal cellular health. The inability to adequately quench ROS contributes to pathology associated with many neuromuscular and cardiovascular pathologies (Wallace, 2000). Quercetin has been demonstrated to have inherent free radical quenching ability in both in vitro and in vivo models (Nabavi et al., 2012b; Nabavi et al., 2015). Indeed, quercetin appears to directly scavenge free radicals particularly by reaction with attached hydroxyl (OH) groups and inhibition of metal chelation (Afanas' ev et al., 1989). Furthermore, quercetin decreases H2O2 formation through direct inhibition of xanthine oxidase and stimulates the synthesis of important antioxidant enzymes (i.e. superoxide dismutase (SOD), glutathione peroxidase (GPX)) (Chang et al., 1993; Ballmann et al., 2016; Ballmann et al., 2017). Changes in antioxidant enzyme expression with quercetin administration are likely due to induction of PGC-1α or p38 mitogen-activated protein kinase (MAPK) leading to activation of transcription factors (i.e. Nrf-2) important in controlling antioxidant enzyme expression (Li et al., 2016). Uncoupling of endothelial nitric oxide synthase (eNOS) appears to be a primary source of endothelial-dependent free radical production (Ding et al., 2007). In the context of CVD, this may lead to microvascular dysfunction and impaired vasodilatory control. Quercetin has been shown to decrease eNOS uncoupling and prevents superoxide formation in vascular muscle in rat aortic rings (Romero et al., 2009). Quercetin-mediated inhibition of endothelin-1 (ET-1) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity are likely contributors to prevention of free radical accumulation and maintenance of vascular function.
In skeletal muscle, quercetin has been shown to inhibit lipid peroxidation and increase antioxidant enzyme activities (Ekinci Akdemir et al., 2016). Additionally, quercetin supplementation has been reported to decrease DNA damage and preserve antioxidant function in skeletal muscle of diabetic mice (Alam et al., 2014). In the heart, quercetin similarly decreases lipid peroxidation while also restoring glutathione levels during pro-oxidative states (Nabavi et al., 2012a). Attenuation of cardiac remodeling and heart failure has been documented with quercetin-mediated increases in cardiac SOD2 and GPX2 (Ballmann et al., 2016; Ballmann et al., 2017). Taken together, a promising effect of quercetin therapy in cardiac and neuromuscular diseases is through prevention of oxidative stress via inherent antioxidant properties of quercetin and changes in antioxidant enzyme physiology, such as through inhibition of ET-1-induced p47phox overexpression and NADPH oxidase overactivity.
Autophagy and Apoptosis
Autophagy is a catabolic process initiated to maintain cellular health by removal of damaged or surplus components via lysosomal degradation. Under normal physiological conditions, autophagy maintains proper cellular metabolism and degraded constituents can be recycled and input into biochemical processes thus promoting cell survival (Rabinowitz and White, 2010). Autophagic flux has a fine balance between physiological and pathological states whereby too little or too much autophagic activity in striated muscle can contribute to pathology associated with both CVD and NMD (Martinet et al., 2007; Carnio et al., 2014).
Initiation of autophagy is a complex process leading to the formation of autophagosomes and lysosomal degradation, which is modulated by damage to cellular constituents, oxidative stress, inflammatory signaling, and mitochondrial dysfunction (Filomeni et al., 2015; Lapaquette et al., 2015). Autophagy, in part, regulates cell death through cross-talk of similar signaling transducers (i.e. p53, c-Jun N-terminal kinase, Bak) also involved in apoptosis (Marino et al., 2014). Physiologically, apoptosis is necessary for fetal development and maintenance of healthy tissue through the regulation of death of dysfunctional cells (Reed, 2000). Like autophagy, pro-apoptotic signaling is tightly controlled as failure to induce apoptosis in dysfunctional cells ultimately leads to poor tissue health and hyperactive signaling can lead to death of viable tissue. Apoptotic processes play a pivotal role in development of pathologies in cardiac and skeletal muscle (Tews, 2005; Lee and Gustafsson, 2009). Signaling cascades of apoptosis in striated muscle can be induced by free radical formation, DNA fragmentation, cytokine signaling, and mitochondrial damage (Mayer and Oberbauer, 2003; Andrianjafiniony et al., 2010). Since increased mitochondrial dysfunction, oxidative stress, and inflammation are commonly associated with CVD and NMD, it is not surprising that autophagy and apoptosis mediate, at least in part, both development and severity of these conditions. Potentially maladaptive autophagic and apoptotic responses have been implicated in dystrophic heart failure (Spaulding et al., 2019b), ischemia-reperfusion (IR) injury (Quindry et al., 2005), sarcopenia/disuse atrophy (Tews, 2002), and Duchenne muscular dystrophy (DMD) (Spaulding et al., 2018) among other pathologies.
Numerous investigations indicate that quercetin treatment is associated with apparent modulation of maladaptive autophagic and apoptotic signaling. Protective autophagy has been demonstrated to be induced by quercetin treatment in a variety of in vitro models (Cui et al., 2015; He et al., 2017), likely due to increased calcium release (Cui et al., 2015). Additionally, quercetin is protective against IR injury by inducing autophagy in in vivo models through AMPK-dependent mechanisms (Chen et al., 2014). Inhibition of fibroblast proliferation and fibrotic tissue deposition is attenuated and mediated by quercetin through maintenance of adaptative autophagy (Lv et al., 2017). Quercetin differentially affects apoptotic signaling based on cell type and microenvironment. In cancer cell models, quercetin is pro-apoptotic through caspase and p38 activation (Chien et al., 2009; Lee et al., 2010). In this context, quercetin stimulated apoptosis is interpreted to be adaptive as induction of apoptosis to uncontrolled cell growth is a generally desirable outcome. In dystrophic heart disease, quercetin does not appear to alter autophagy related protein expression or markers of autophagosome formation (Quindry et al., 2017). This has also been noted in dystrophic skeletal muscle indicating that autophagy modulation may be tissue and disease specific.
In CVD and NMD, quercetin has been shown to be anti-apoptotic leading to increased viability of tissue. In vitro quercetin treatment improves cardiomyocyte survival following IR challenge through inhibition of oxidative stress and apoptosis (Shu et al., 2019). Additionally, quercetin decreased apoptotic cell death in rodent hearts following IR injury through activation of PI3K/Akt (Liu et al., 2014). Protection against free radical-mediated mitochondrial dysfunction has been implicated in quercetin’s anti-apoptotic actions (Park et al., 2003). Quercetin supplementation also exerts anti-apoptotic effects in glycolytic skeletal muscle during statin use by decreasing mitochondrial free radial damage (Bouitbir et al., 2016). While quercetin differentially affects autophagy and apoptosis, growing consensus suggests adaptive response to treatment making it a viable therapy for a variety of CVD and NMDs with maladaptive responses.
Cardiovascular conditions
Coronary Heart Disease (CHD)
Cardiovascular diseases are the leading cause of death worldwide with almost half of all cardiovascular related events being attributed to CHD. Indeed, 1 out of 7 deaths in the US in 2013 were attributed to CHD (Mozaffarian et al., 2016). The typical physiological underpinning of CHD is the formation of atherosclerotic plaques, which causes narrowing of coronary vessels, and subsequent myocardial ischemia. Plaque formation is driven, in part, by inflammatory responses and in serious cases, plaques may become destabilized and cause thrombotic events in other tissues (Libby et al., 2002). While non-modifiable risk factors such as age, sex, and race contribute to development of CHD, controlling modifiable risk factors such as smoking, physical inactivity, and poor dietary habits are among the most potent prevention strategies of CHD. Intriguingly, quercetin supplementation has been reported to induce molecular pathways in cardiac tissue typically associated with adaptations to exercise and caloric restriction, which may provide a positive impact on CHD prevention (Ballmann et al., 2016; Patel et al., 2018). Flavonol consumption, including quercetin, is associated with an improved cardiovascular risk factor profile and lower CHD mortality (Lin et al., 2007). Quercetin administration improves coronary vessel vasodilatory responses in an endothelial-independent fashion suggesting quercetin may improve myocardial blood flow (Rendig et al., 2001). Dyslipidemia is a contributing risk factor for atherosclerosis and CHD but is also responsive to the introduction of healthy lifestyle behaviors such as regular physical activity and appropriate dietary choices. Regular quercetin consumption in the form of high concentration dietary supplements is associated with decreased plasma triglycerides, LDL, cholesterol, and increased high-density lipoprotein in dyslipidemic patients (Talirevic and Sehovic, 2012). As previously mentioned, quercetin-dependent inhibition of MMP-1 prevents atherosclerotic plaque formation and destabilization by blunting LDL oxidation (Song et al., 2001). Recent evidence in patients with CHD indicates that quercetin supplementation decreased inflammation by inhibiting transcriptional activity in mononuclear cells, which contribute to plaque formation (Chekalina et al., 2018). While quercetin supplementation is not a substitute for healthy lifestyle choices, collective evidence suggests that quercetin consumption may aid in decreasing risk factors, development, and complications associated with CHD.
Hypertension/High blood pressure (HBP)
Hypertension (HBP), defined as systolic blood pressure ≥140 and diastolic blood pressure ≥90 for the purposes of this review, affects nearly 33% of all US adults over the age of 20. Compared to other cardiovascular risk factors, HBP is the leading cause of death among risk factors in women and second leading cause in men (Mozaffarian et al., 2016). While lifestyle modifications (i.e. diet, exercise) are often first line treatment of HBP, many cases require pharmaceutical intervention, which are typically successful but can have high inter-individual variation in efficacy (Ruilope, 2012). Unfortunately, antihypertensive medication adherence is low and attitudes toward prescription therapy can be poor. Evidence has shown that non-adhering patients have reported wishing to eliminate the need for prescription medication and find more holistic approaches (Jolles et al., 2013).
Naturally occurring quercetin and conjugates have been shown to have antihypertensive effects through various mechanisms including decreased angiotensin converting enzyme (ACE) activity, ROS scavenging, and improved vascular smooth muscle function (Larson et al., 2010). Oral and intravenous administration of quercetin potently inhibits ACE activity to a similar extent as captopril, a common antihypertensive pharmaceutical. Additionally, quercetin therapy is associated with decreased responses to angiotensin I and serves as a synergist for bradykinin-mediated modulation of blood pressure (Häckl et al., 2002). ACE protein and mRNA expression are also found to be lower in conjunction with quercetin treatment (Parichatikanond et al., 2012). Given this, quercetin supplementation in substitute or in conjunction with lower doses of ACE inhibitors is a desirable clinical application as adverse respiratory symptoms may be associated with moderate to high doses of ACE inhibitors (Pinargote et al., 2014). Since quercetin possesses direct and indirect antioxidant properties, and enhancement of endogenous antioxidant enzymatic function, quercetin has been postulated to combat HBP through attenuation of vascular dysfunction via ROS scavenging. In spontaneously hypertensive rats, daily oral quercetin supplementation decreases blood pressure, urinary isoprotanes, and plasma malonyldialdehyde (MDA) concentration (Duarte et al., 2001). In hypertensive humans quercetin corrects HBP independent of an impact on oxidative stress (Edwards et al., 2007; Larson et al., 2010). Furthermore, quercetin appears to improve vessel function, which may improve HBP, via activation of AMPK signaling and increased eNOS-mediated nitric oxide (NO) production (Shen et al., 2012). Of particular note, protection is lost with AMPK blockade. Furthermore, acute treatment of quercetin enhances relaxation of aortic smooth muscle in part through increased NO production via activation of eNOS. Treatment with L-NAME, a potent NO inhibitor, abolishes quercetin induced relaxation suggesting that increases in eNOS activity and NO bioavailability are necessary for protective vascular effects (Khoo et al., 2010). Hence, while the importance of the antioxidant action of quercetin is unclear in this context, evidence overwhelmingly indicates that quercetin modulates ACE and improves vascular function thereby conferring protection against HBP. Although molecular mechanisms for lowering of blood pressure with quercetin in humans are not fully understood, systematic review of control trials revealed potent anti-hypertensive effects as measured though clinical outcomes (Serban et al., 2016). Thus, lowering of HBP in humans appears to be feasible with quercetin supplementation but more studies are needed to dissect particular molecular mechanisms involved.
Ischemia-reperfusion (IR) injury- Heart
As previously mentioned, CHD is the leading cause of morbidity and mortality both in the United States and globally (Mozaffarian et al., 2016). Mature progression of CHD eventually leads to occlusion of coronary vessels causing ischemic insult to myocardial tissue resulting in arrhythmias, calcium overload, oxidative stress, and loss of systolic contractile function (Powers et al., 2008). Rapid reestablishment of blood flow in less than 20 minutes can prevent irreversible tissue damage but causes temporary reductions in cardiac function, aptly termed ‘myocardial stunning’ (Bolli, 1992). Longer durations of ischemia lead to irreversible necrotic and apoptotic cell death (i.e. myocardial infarction) due to further calcium overload, oxidative stress, protease activation, and leukocyte infiltration (Powers et al., 2008). Timely reperfusion of blood flow to the myocardium is necessary for survival but induces signaling cascades that exacerbate tissue damage leading to the phenomenon of IR injury. Indeed, reperfusion worsens previously mentioned maladaptive signaling resulting from ischemia and increases ionic disturbances, enzyme denaturation, and mitochondrial dysfunction (Hausenloy and Yellon, 2013).
To prevent and/or decrease severity in cardiac IR injury in the event of an insult (i.e. preconditioning), both pharmaceutical and lifestyle modification (i.e. diet, exercise) interventions have been implicated in providing protection. Among lifestyle modifications, physical activity and exercise lead to robust preconditioning against IR-injury through improved myocardial antioxidant capacity, heat shock protein activation, improved calcium handling, and induction of cellular pathways involved in metabolic homeostasis (i.e. AMPK, SIRT1/PGC-1α, AKT) (Vega et al., 2017). Growing evidence indicates that regular quercetin consumption is cardioprotective against IR injury through induction of phenotypes similar to exercise (Ferenczyova et al., 2020). Cardiomyocytes incubated with quercetin aglycone prior to hypoxia/reoxygenation resulted in improved cell survival through induction of SIRT1/PGC-1α (Tang et al., 2019). Cardiomyoblast (H9C2) cells incubated in quercetin derivatives in vitro exhibit decreased oxidative stress and apoptotic cell death in response to hypoxia/reoxygenation via a PI3K/Akt-dependent mechanism (Shu et al., 2019). Interestingly, quercetin-mediated benefits through PI3K/Akt following IR are also possible through post-conditioning and attenuate pathologic apoptotic cell death (Wang et al., 2013). Quercetin mediated inhibition of mitochondrial permeability transition pore (mPTP) opening may also attenuate mitochondrial calcium overload and mitochondrial dysfunction during IR challenge (De Marchi et al., 2009).
These improvements in parallel mechanisms of mitochondrial function are bolstered by in vivo evidence showing cardioprotective effects of dietary quercetin administration. Pre-treatment with intravenous quercetin (1 mg/kg) reduced infarct size, preserved cardiac function, and prevented inflammation through decreased cytokine signaling during acute IR (Jin et al., 2012). Acute and chronic quercetin regimens appear to protect myocardial function from global IR during isolated working heart preparations supported by underlying decreases in markers of tissue damage (i.e. creatine kinase) and lipid peroxidation (Ikizler et al., 2007). Fourteen days of quercetin enrichment, albeit in conjunction with α- tocopherol, increased ATPase activity and decreased lysosomal enzyme fractions during post-IR suggesting an anti-catabolic state in the treated myocardium (Punithavathi and Prince, 2010).
While confirmatory evidence in humans regarding IR injury is lacking (due to ethical considerations), indirect measures demonstrating a quercetin-mediated prevention or decreased severity of IR injury have been documented. Diets higher in flavonol consumption, including quercetin, decrease CVD risk and increase longevity (Perez-Vizcaino and Duarte, 2010). Quercetin enrichment decreased human platelet aggregation and eicosanoid formation suggesting that quercetin may reduce IR injury by decreasing the risk of a thrombolytic event in the coronary arteries (Pace-Asciak et al., 1995). In patients with stable CHD, short-term adjunctive therapy with quercetin (120 mg/day) while undergoing 24-hour Holter ECG monitoring had a significantly lower number of premature ventricular contractions and ST-segment depression episodes of which the latter suggests improvements in coronary flow (Chekalina et al., 2018). However, disparities exist as others have shown that despite dose dependent increases in plasma quercetin levels, little or no improvements in vascular function were found following quercetin supplementation in humans (Bondonno et al., 2016; Brüll et al., 2017). However, it should be mentioned that these investigations used healthy men and women making it difficult to detect difference in variations from already normal vascular function. This is supported by other investigations showing lack of efficacy of quercetin supplementation in healthy subject suggesting that quercetin benefits may manifest themselves in disease populations (Conquer et al., 1998). Thus, it is possible that there may be a limitation in the therapeutic potential of quercetin in near healthy or very mild phenotypes.
Overall, in vitro and in vivo models serves as convincing data that quercetin consumption is linked to a series of beneficial cellular mechanistic alterations for IR injury resilience. There is currently insufficient data to formally conclude that quercetin attenuates IR injury in the myocardium, however, currently available data are promising. Development of an anti-thrombolytic reperfusion “cocktail therapy” including quercetin may be feasible in terms of treatment but more controlled human trials are needed to understand specific benefits.
Dystrophic Heart Failure (DHF)
DMD is an X-linked genetic disease, which impacts approximately 1 in every 3,500-5,000 male births causing functional loss of the protein dystrophin (Kunkel et al., 1989). Dystrophin is important in maintaining cellular membrane integrity during muscular contraction and loss of dystrophin causes progressive heart failure. DMD causes pre-mature mortality and approximately 40% of all deaths of DMD patients are due to cardiac complications (Lamperti and Moggio, 2010). Underlying pathology of DHF, secondary to dystrophin deficiency, is multi-faceted and includes increased oxidative stress, metabolic dysfunction, cardiac remodeling, and fibrosis, among others (Rahimov and Kunkel, 2013). Thus, a countermeasure that can attenuate all or multiple secondary pathological underpinnings is a clinical imperative.
Recent investigations in dystrophic mouse models (i.e. mdx, mdx/utrn+/-) provide preliminary evidence that dietary quercetin enrichment can both rescue developed DHF and prevent it (Ballmann et al., 2015; Ballmann et al., 2016; Ballmann et al., 2017). In mdx mice with advanced disease progression, treatment with long-term dietary quercetin attenuated pathological increases in heart weight and decreased cardiac damage (Ballmann et al., 2015). Improvements in DHF were attributed, in part, to lower levels of transforming growth factor-α1 (TGF-α1), which is a key mediator in fibrotic tissue deposition. In recent preventative studies, life-long dietary quercetin supplementation has been shown to have robust benefits in multiple facets of DHF. In mdx and mdx/utrn+/- mice, life-long quercetin administration increased myocardial PGC-1α protein abundance and improved associated outcomes including increased expression of mitochondrial and antioxidant enzymes (Ballmann et al., 2016; Ballmann et al., 2017). Importantly, these findings may reflect increased myocardial antioxidant capacity and improved metabolic function. Furthermore, markers of cardiac remodeling (i.e MMP-9), fibrosis (i.e fibronectin), and myocardial damage were decreased. Life-long quercetin fed mdx/utrn+/- mice also displayed decreased cardiac edema and improved cardiac function (Ballmann et al., 2016).
A promising therapy in DHF is increasing expression of utrophin, a dystrophin-like protein that may be able to functionally replace dystrophin in its absence. Remarkably, life-long quercetin treatment increased cardiac utrophin expression in mdx and mdx/utrn+/- mice and appears to result in reassembly of the dystrophin-glycoprotein complex (DGC) (Ballmann et al., 2016; Ballmann et al., 2017). Since mdx/utrn+/- mice already have a partial loss of utrophin, these findings suggest quercetin has a potent effect on utrophin regulation. Likely a result of these adaptations, end-stage measurement of physical activity in quercetin- fed mice was enhanced suggesting improvements in functional capacity (Ballmann et al., 2016; Ballmann et al., 2017). Altogether, quercetin enrichment appears to treat and prevent DHF by inducing adaptations in pivotal underlying pathological mechanisms, which ultimately led to apparent improvements in cardiac health and functional capacity. However, it should be cautioned that not all investigations have supported chronic use of quercetin in the progression of dystrophic heart failure and no studies to date have investigated how these findings translate to humans. Disparities between these findings are likely due to severity of phenotype whereby more advanced or severe disease progression may be more resistant to quercetin therapy. In particular, this may be due to the fact that underlying DHF is a functional loss of dystrophin by which quercetin therapy, among others human treatments, will not be able to ameliorate. As such, it is worth noting that the findings summarized within are observed within relatively mild forms of the disease, which manifest as systolic dysfunction. Accordingly, more severe forms of DHF, in other mouse models – not to mention other species – and those with diastolic dysfunction may not exhibit a comparable responsiveness to quercetin. Thus, quercetin treatment for dystrophic heart failure should be approached with optimism but not certainty. Given evidence of improved cardiovascular function with quercetin enrichment in other CVDs, quercetin therapy should be tested through randomized control trails to determine if benefits seen in other pathologies translate to DHF.
Doxorubicin Cardiotoxicity
Each year in the United States alone, approximately 650,000 patients undergo chemotherapy treatment (Dunbar et al., 2014). Of the various chemotherapy agents, doxorubicin is one of the most common treatments used to treat a wide arrange of cancers including breast, kidney, thyroid, lung, bone, nerve tissue, joint, and soft tissue types. Anti-cancer effects are due to intercalation in, and chelation of DNA and subsequent inhibition of topoisomerase II and arrest of replication (Tacar et al., 2013). Doxorubicin treatment also results in high levels of free radical production when the compound is converted to the semiquinone form in mitochondria. Since cardiomyocytes are mitochondria-dense and particularly vulnerable to free radical damage, doxorubicin is considered cardiotoxic and often leads to dilated cardiomyopathy. Incidence of cardiovascular side effects are dose dependent and higher doses are more likely to cause congestive heart failure (Chatterjee et al., 2010). As previously mentioned, quercetin has inherent antioxidant properties and in therapeutic doses is linked to increased endogenous antioxidant enzyme production in the heart, making it an appealing preventative measure to doxorubicin cardiotoxicity. Indeed, quercetin appears to attenuate the negative effects of doxorubicin through in vitro and in vivo models (Dong et al., 2014b).
To this end, quercetin treatment was closely linked to elevated SOD2 expression and decrease doxorubicin-induced lipid peroxidation in neonatal cardiomyocytes (Dong et al., 2014b). Furthermore, quercetin increased cell viability and cytoskeletal proteins important for cellular repair following doxorubicin treatment in cardiomyocytes (Chen et al., 2013). Since cardiomyocytes have limited regeneration potential, improvements in cell survival and repair are critical and could mediate quercetin protection against myocardial death during treatment. The application of quercetin is also tied to the blunting of doxorubicin-induced arrhythmias and reduces myocardial remodeling through inhibition of apoptotic cell death in rat hearts (Pei et al., 2007). Recent in vivo experiments indicate that quercetin enrichment decreases biomarkers of myocardial damage, increases antioxidant enzyme activity, lowers tissue lipid peroxidation, and blunts DNA damage following doxorubicin therapy in mice (Zakaria et al., 2018). Mechanistically, protection was likely due to quercetin-mediated elevations of AMPK and PGC-1α expression, which regulate the oxidative phenotype, metabolic adaptations, and antioxidant enzyme function. Similar findings are reported in other organ systems and human cell lines (Jambhulkar et al., 2014). While these early results are promising, we are unaware of studies investigating quercetin efficacy in preventing doxorubicin-associated cardiac side effects in humans. Quercetin appears to have therapeutic potential to decrease myocardial damage and improve cardiac metabolism with doxorubicin administration, making it a potentially viable adjunctive therapy during cancer treatment.
Neuromuscular conditions
Ischemia-reperfusion (IR) injury- Muscle
Insufficient blood flow and oxygen delivery, particularly during increased demands created during even low intensity exercise like walking, can cause skeletal muscle to experience periodic ischemic events. Ischemic insult to skeletal muscle causes perturbations in metabolism impairing phosphagen and glycogen re-synthesis (Harris et al., 1986), as well as mitochondrial health (Pipinos et al., 2006). Long-term ischemia can result in tissue damage and in extreme cases, necrotic cell death. Reestablishing adequate blood flow is necessary to avoid irreversible tissue damage. However, rapid restoration of blood flow, or reperfusion, can also cause maladaptive responses influencing susceptibility to tissue injury, inflammation, vascular function, and oxidative stress (Gute et al., 1998). IR injury occurs at the local site of insult but can have systemic effects leading to injury of other tissues (Blaisdell, 2002). Skeletal muscle IR injury is a multifaceted pathological process affecting numerous types of tissues, cells, and biochemical pathways.
In a series of experimental contexts, quercetin has been repeatedly shown to mitigate negative responses to skeletal muscle IR injury. For example, in independent experiments, rats and rabbits supplemented with oral quercetin each demonstrated increased antioxidant enzyme activities and decreased free radical injury following IR (Ekinci Akdemir et al., 2016). Furthermore, in these investigations rabbits supplemented with quercetin exhibited lower superoxide concentrations in skeletal muscle post- IR insult (Huk et al., 1998). Dietary quercetin glycoside administration improved skeletal muscle angiogenic responses to ischemic insult in an eNOS dependent fashion (Sumi et al., 2013). Supporting this, NO concentrations in response to IR are reported to be higher with quercetin supplementation suggesting improved vasodilatory control (Huk et al., 1998). By extension, recent data in humans treated with quercetin-containing supplements also suggest skeletal muscle contractile function is preserved in response to an IR challenge following IR (Gelabert-Rebato et al., 2018). Skeletal muscle O2 extraction is also improved following acute IR in quercetin fed men and women with concurrent increases in anaerobic exercise performance (Gelabert-Rebato et al., 2019). Mediation of many these benefits are likely through SIRT1/PGC-1α by improving capillary density and antioxidant enzyme expression. Thus, quercetin appears to improve oxidative stress and vascular outcomes following IR insult while also preserving contractile function of skeletal muscle. From a clinical perspective, quercetin treatment alone or in conjunction with conventional treatments could be potentially applicable to pathologies such as peripheral vascular disease or renal artery disease where vasodilatory control is poor.
Duchenne muscular dystrophy (DMD)- Locomotor muscle
As previously mentioned, DMD is caused by the absence of the cytoskeletal protein dystrophin. Loss of dystrophin leads to membrane fragility, and dystrophic muscle is particularly sensitive to contraction-induced injury (Ervasti and Campbell, 1993). Dystrophic locomotor muscle progressively loses the ability to produce force resulting in poor balance, altered gait, fatigue, and loss of strength (Emery et al., 2015). Affected patients often are wheelchair bound and lose the ability to ambulate by adolescence or earlier. Exercise and physical activity have been shown to preserve ambulation and benefit dystrophic skeletal muscle (Grange and Call, 2007), though this is not without controversy as numerous reports indicate accelerated disease processes with exercise (Selsby et al., 2013; Spaulding and Selsby, 2018). Thus, stimulating exercise-induced pathways are a promising avenue to improve both quality and quantity of life in DMD patients.
Since quercetin can mediate SIRT1/PGC-1α activation in addition to possessing inherent antioxidant and anti-inflammatory properties, it is a good candidate to serve as a natural therapeutic in DMD. Long-term dietary quercetin enrichment has been shown to improve dystrophic skeletal muscle function in mdx mice, but only partially (Spaulding et al., 2016b). After 12 months of quercetin supplementation, specific tension and resistance to fatigue were preserved in slow twitch hind-limb muscle (i.e. soleus). However, these functional improvements were not detected in primarily fast twitch muscle (i.e. extensor digitorum longus (EDL)) suggesting that benefits could possibly be mediated by fiber type. Post-mortem histological analysis revealed similar muscle injury and fibrosis in quercetin-treated and untreated animals (Spaulding et al., 2016b). Interestingly, quercetin preserved locomotor activity at end-stages of the mdx mice suggesting that even modest protection of skeletal muscle may preserve ambulatory ability. However, quercetin has also been reported to induce neural excitation due to adenosine antagonism in the brain (Alexander, 2006). Whether these activity changes are fully due to maintenance of skeletal muscle health or purported neural excitation due to adenosine antagonism in the brain remains unclear at this time.
The mdx mouse model has a relatively mild phenotype compared to dystrophin deficiency in humans, hence recent follow up studies using mdx/utrn+/- and D2-mdx mice, which have more severe phenotypes, have been conducted. Dietary quercetin supplementation did not maintain skeletal muscle function in mdx/utrn+/- mice (Selsby et al., unpublished observations). Further, 7 months of quercetin administration did not improve skeletal muscle function or decrease histological parameters of disease in D2-mdx mice regardless of muscle or fiber-type (Spaulding et al., 2019a). Of interest, quercetin was used in combination with nicotinamide riboside, Lisinopril, and/or prednisolone and also failed to attenuate disease severity (Spaulding et al., 2019a). Thus, disease severity may influence efficacy of quercetin administration in dystrophic muscle. To date, functional analysis and histology of dystrophic locomotor muscle have only been studied after long-term supplementation at end-stage time points. The extent to which quercetin supplementation improves outcomes earlier in the therapy time course for dystrophic muscle is unknown. Quercetin appears to impose some benefit to dystrophic muscle in some models but serial measurements during treatment are needed to elucidate therapeutic benefit. However, given the data collected to date we expect quercetin’s capacity to attenuate disease severity to be limited as human disease presentation aligns more closely with the phenotypes of mdx/utrn+/- and D2-mdx mice. This may be especially true when disease progression has advanced and functional capacity is lost in humans. Given the collective evidence in animals, clinical application of quercetin in DMD is limited leaving the need for further human study in conjunction with existing therapies. Conversely, more promising results in clinical human trials have been found using epigallocatechin gallate, a similar compound.
Duchenne Muscular Dystrophy (DMD)- Respiratory muscle
Diaphragm and respiratory muscle failure leads to premature mortality in many DMD patients (Eagle et al., 2002). Like in locomotor muscle, dystrophin deficiency leads to increased oxidative stress, metabolic dysfunction, fibrosis, and necrotic tissue damage (Petrof, 1998). Improvements in secondary care, primarily through respiratory support therapy, have extended average lifespan of DMD patients. However, prolonged mechanical ventilation may accelerate diaphragm muscle decline eventually hastening the need for full-time mechanical ventilation use (Baydun et al., 1990). Thus, continued development of pragmatic therapies to attenuate respiratory muscle dysfunction in DMD are needed.
In an early experiment, oral quercetin delivery decreased histopathological injury (i.e. muscle necrosis, fibrotic protein expression) in diaphragms from mdx mice using a rescue treatment strategy (3-9 mo of age) (Hollinger et al., 2015). While mechanisms underlying these findings are not fully clear, quercetin-mediated PGC-1α activation appears likely, especially considering similar adaptive responses with adeno-associated virus (AAV) driven PGC-1α overexpression (Hollinger et al., 2013).
A follow up study conducted in mdx mice with long-term quercetin supplementation included serial measurements of respiratory function (Selsby et al., 2016). Tidal volume in quercetin-treated animals was preserved up to 10 months of age while peak inspiratory and expiratory flow was maintained up to 6 months of age. However, end-point respiratory function analysis showed similar levels of decline in mdx mice regardless of quercetin administration (Selsby et al., 2016). Consistent with the loss of respiratory function protection at the end time point (14 months of age), post-mortem analysis of diaphragm muscle did not support quercetin therapeutic efficacy. Fibrosis and impaired functional parameters were similar regardless of quercetin enrichment. In addition, many adaptive genes involved in metabolism and mitochondrial biogenesis were suppressed in mdx mice compared to healthy mice, independent of quercetin enrichment. This long-term quercetin insensitivity in respiratory muscle is consistent with previous findings in limb skeletal muscle of dystrophic animals (Spaulding et al., 2019a) and more recent work in the severe D2-mdx model (Spaulding et al., 2020). The addition of nicotinamide riboside, Lisinopril, and/or prednisolone also failed to protect respiratory function and diaphragms in dystrophic muscle. These findings may in part be due to lack of sustained pathway activation with quercetin treatment. Previously mentioned studies showing favorable disease outcomes in respiratory muscle were observed earlier in the disease progression whereas more recent evidence suggests this is lost over time. Other investigations assessing cardiac outcomes at later timepoints showed more favorable results, but this is likely due to greater diaphragmatic injury by 14 months in dystrophic mouse models compared to the myocardium. Given the failure to sustain a therapeutic effect, use of quercetin to offset impaired respiratory function in DMD is not warranted at this time and may not be a viable option for treatment.
Disuse and Atrophy
Skeletal muscle responds to disuse and decreased activation through catabolic mediators that cause atrophy (Bodine et al., 2001). Muscle atrophy is a functional consequence of multiple disease states (i.e. cancer, diabetes), chronic bed rest, denervation, and aging. Underlying these conditions are complex signaling pathways responsible for catabolic cellular responses. While heterogeneity for atrophic signals between conditions exists, most types of skeletal muscle atrophy have a common transcriptional signature. Transcripts involved in important processes including ATP production, inflammation, and protein degradation have been shown to be altered in most forms of muscle atrophy (Lecker et al., 2004). Through the mediation of these processes, quercetin supplementation may blunt atrophy that accompanies a variety of conditions. In cachexic muscle wasting, grip strength and muscle mass was preserved in mice with oral quercetin feeding (Velázquez et al., 2014). In disuse models, dietary quercetin caused PGC-1α-mediated reduction in mitochondrial ROS formation, increased mitochondrial enzyme expression, and suppressed AKT protein degradation (Mukai et al., 2016). Ubiquitin ligase-induced protein degradation in skeletal muscle was blunted with quercetin administration in a disuse model (Mukai et al., 2010). Recently, the inflammatory cytokine TNF-α was shown to be decreased with quercetin administration resulting in suppression of atrophic factors and markers of inflammatory signaling in mice (Kim et al., 2018). Supporting this, quercetin glycoside dietary enrichment attenuated loss in skeletal muscle mass and expression of atrophic gene expression with dexamethasone treatment (Otsuka et al., 2019). Collectively, these findings suggest that quercetin suppresses atrophic signaling, inflammation, and protein degradation during disuse of skeletal muscle resulting in preserved muscle mass and function. While previous evidence in animal models is compelling, we are unaware of human studies demonstrating these principles. Given the burden of muscle atrophy with various chronic diseases, we identify this as an urgent need for clinical investigation.
Muscle Damage and Inflammation
Muscular damage can occur as an acute insult (i.e. overload, exercise) or result of chronic disease or pathology (i.e. muscular dystrophy, diabetes, etc.). With damage, an accompanying inflammatory response occurs hours to days post-injury in order to facilitate removal of damaged tissue and cellular repair (Tiidus, 2010). Indeed, immune cell infiltration increases in injured sites of skeletal muscle with oxidative burst and phagocytotic mechanisms aiding in removal of damaged cellular debris (Tiidus, 2008). While inflammatory responses following muscle damage appears necessary for successful repair and effective adaption, non-specific damage can occur to nearby healthy tissue exacerbating injury and soreness (Brunelli and Rovere-Querini, 2008). Chronic inflammation associated with a number of disease states appears to contribute to disease progression and pathological remodeling of muscle tissue (Hollinger et al., 2015; Spaulding et al., 2016a). Thus, mediating damage and inflammatory processes is needed to maintain skeletal muscle health and function. Quercetin mediates damage and anti-inflammatory responses in animals and humans, but benefits are unclear. Life-long quercetin enrichment preserved functional capacity and specific force of fibrotic and damaged soleus muscle in dystrophic mice (Spaulding et al., 2016a). In dystrophin-deficient muscle and muscle from diabetic mice, long-term quercetin administration decreased inflammation and expression of inflammatory mediators while also slowing disease progression (Hollinger et al., 2015). In diabetic mice, quercetin feeding reduced inflammatory biomarkers (i.e. TNF-α, inducible NOS) while aiding in maintenance of skeletal muscle glucose metabolism (Anhê et al., 2012).
While previous studies support some benefits of quercetin supplementation on chronic disease and skeletal muscle damage, evidence of quercetin-mediated protection from acute muscle damage is equivocal. Quercetin prevention of acute muscle damage and inflammation has been primarily studied within the context of eccentric overload or intense exercise. Eight-weeks of 1,000 mg/day quercetin supplementation did not alter plasma creatine kinase, lactate dehydrogenase, or other markers of muscle damage in male athletes (Daneshvar et al., 2013). Also, dietary quercetin administered 7 days before and 5 days after acute eccentric muscle injury did not attenuate soreness or plasma markers of muscle damage (O’Fallon et al., 2012). Conversely, a recent investigation reported preserved isometric strength, decreased plasma biomarkers of muscle damage, and lower muscle soreness with 14 days of quercetin treatment (1,000 mg/day) (Bazzucchi et al., 2019). Complicating interpretation of these data, however, is the variety of injury-inducing exercise protocols, which differed by intensity and volume/load. Furthermore, dosing protocols (low vs. high) and quercetin forms (aglycone vs. glycone) also differed. Thus, we highlight this as an area with need for continued study with standardized muscle injury protocols and dosing regimens.
Conclusions, Clinical implications, and Future directions
Quercetin is a known antioxidant and anti-inflammatory that also activates molecular modulators of skeletal muscle and cardiac muscle metabolic adaption, extracellular matrix remodeling, and oxidative stress. Specifically, quercetin stimulates AMPK activity resulting in increased mitochondrial biogenesis, improved mitochondrial quality, increased autophagy, GLUT4 translocation, and glycogen synthesis. PGC-1α and SIRT1, downstream of AMPK, are similarly increased by quercetin and stimulate mitochondrial biogenesis and increased antioxidant capacity. Quercetin can also alter expression of MMPs such that MMP1 was upregulated to help maintain plaque stability in atherosclerosis and MMP2 and MMP9 were reduced to attenuate cardiac remodeling and preserve cardiac function in dystrophic cardiac pathology. Commonly, CVD and NMD exhibit mitochondrial dysfunction, oxidative stress, and inflammation that contribute to disease pathologies. As a nutraceutical capable of reducing oxidative stress, decreasing inflammation, and stimulating molecular mechanisms to combat mitochondrial dysfunction and detrimental cardiac remodeling, quercetin has the potential to positively impact aspects of cardiac and skeletal muscle health in a variety of pathologies (Figure 3). However, it should be noted that in some pathologies, quercetin treatment produced trivial or uncertain effects. Thus, further study is needed to confirm whether the various diseased populations discussed currently may benefit therapeutically from quercetin supplementation.

In a new window | Download PPT
Figure 3: Quercetin effects on skeletal and cardiac muscle in CVD and NMD. Summary of possible benefits and mechanisms by which quercetin may confer protection against various chronic diseases. Uncertain outcomes are followed by “?”.
The clinical implications of quercetin treatment are multi-faceted. First, quercetin is widely present in most diets globally and relatively available for consumption. Bioavailability of quercetin has been shown to be higher when consumed through natural dietary sources. Thus, nutritional modifications targeting foods high in quercetin conjugates may provide a practical avenue to impart benefits. However, many investigations utilizing animal models employ supraphysiological doses, while the optimum dose of quercetin in humans has not been established. The feasibility of attaining therapeutic benefits through increasing quercetin solely by natural dietary sources is unknown and should be investigated further. Second, evidence from currently available research indicates that quercetin is safe in most scientific and clinical contexts; resulting in the designation of FDA GRAS status in the early part of the century.
In support of this GRAS designation, quercetin toxicity appears to be low, suggesting that untoward effects due to exogenous supplementation of quercetin are a remote possibility. Given that adherence to therapeutic interventions may be related to negative symptoms, quercetin treatment could provide a safe and adherable therapy to be used in a variety of diseased populations. Lastly and most importantly, quercetin appears to influence multiple underlying pathologic mechanisms both individually and simultaneously. Clinically, this generalized overarching conclusion is important in that the proposed application is made more efficacious by the fact that treatment delivery involves a single therapeutic agent to alleviate a faceted disease. While data suggests that quercetin treatment may not influence every underlying mechanism or disease process to the same extent, the pleiotropic nature of quercetin makes it at least an appealing conjunctive therapy or preventative measure. To fully determine the clinical potential of quercetin, placebo controlled clinical trials in diseased human populations are an essential next step in order to confirm or refute the purported efficacy of quercetin therapy for many chronic conditions.
Indeed, the current review has summarized a wealth of preclinical investigations into the potential efficacy of quercetin as a therapeutic agent against many chronic diseases. While quercetin is not a dietary panacea, we have highlighted a wealth of evidence to suggest that highly concentrated delivery of the compound through dietary supplementation may be of clinical importance. However, given that most of these tentative conclusions are based on pre-clinical investigations in rodent and cell culture studies, transference of findings to humans is not yet warranted. Indeed, findings from animals often do not always translate to humans. Moreover, in this review we have highlighted scenarios for a disease condition where quercetin appeared to benefit one animal paradigm, but was less beneficial, or of no advantage, in a different experimental context. Further tempering the current conclusions, the dosing of quercetin in most of the animal studies detailed in this review are scaled to parallel human applications when normalized for both body weight and surface area. Nonetheless, there is limited evidence suggesting optimal dosage, duration, and efficacy in animals or humans. Extending on this line of reasoning, although short term quercetin supplementation has been reported as safe even at relatively high dosages, it is currently unknown how chronic supplementation might influence safety profiles; a point that applies to healthy populations in addition to those suffering from the chronic conditions outlined in this review. Regardless, quercetin shows promise as an effective therapeutic agent by which even small impacts of quercetin may improve quality of life and clinical end points for a host of chronic diseases.
References
】SChristopher G. Ballmann1
1Department of Kinesiology, Samford University, Birmingham AL.
John C. Quindry2,3
2School of Integrative Physiology and Athletic Training, Montana University, Missoula MT. 3International Heart Institute, St. Patrick’s Hospital, Missoula, MT.
Hannah R. Spaulding4
4Center for Skeletal Muscle Research at Robert M. Berne Cardiovascular Research Center, University of Virginia School of Medicine, Charlottesville, VA.
Joshua T. Selsby5
5Department of Animal Science, Iowa State University, Ames IA.
Corresponding author:
Christopher Ballmann
Email: cballman@samford.edu
In a new window | Download PPT
Figure 1: Chemical structure of quercetin. IUPAC 2- (3,4-dihydroxyphenyl)-3,5,7 trihydroxychromen-4-one. Molecular weight: 302.23 g/mol.
In a new window | Download PPT
Figure 2: Primary quercetin mediated activation of AMPK/SIRT1/PGC-1α. Creation of an acute energy supply demand mismatch activates AMPK and increases intracellular NAD+ concentrations. AMPK leads to phosphorylation and/or the latter results in SIRT1 activation leading to the deacetylation of PGC-1α. Genes involved in mitochondrial biogenesis, oxidative metabolism/antioxidant enzyme production, and slow-twitch genes are upregulated by PGC-1α interaction with nuclear receptors.
In a new window | Download PPT
Figure 3: Quercetin effects on skeletal and cardiac muscle in CVD and NMD. Summary of possible benefits and mechanisms by which quercetin may confer protection against various chronic diseases. Uncertain outcomes are followed by “?”.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 14505 | 63 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA