Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Mitochondrial dynamics and preconditioning in white matter
Time:2018-02-12
Number:8246
Author Affiliations

Conditioning Medicine, 2018. 1(2):64-72.
Abstract
Mechanisms of ischemic preconditioning have been extensively studied in gray matter. However, an ischemic episode affects both the gray matter (GM) and white matter (WM) portions of the brain. Inhibition of mitochondrial fission is one of the mechanisms of preconditioning neuronal cell bodies against ischemia. Although axons are anatomical extensions of neuronal cell bodies, injury mechanisms differ between GM and WM. Indeed, axonal dysfunction is responsible for much of the disability associated with clinical deficits observed after stroke; however, the signaling process underlying preconditioning remains unexplored in axons. Using mouse optic nerve, which is a pure isolated WM tract, we show that mitochondria in myelinated axons undergo rapid and profuse fission during oxygen glucose deprivation (OGD) that is mediated by translocation of cytoplasmic Dynamin Related Protein-1 (Drp-1) to mitochondria. OGD-induced mitochondrial fission correlates with reduced mitochondrial motility and loss of axon function. Mitochondrial fragmentation and loss of motility become permanent during the recovery period. Inhibiting mitochondrial fission by administering mitochondrial division inhibitor-1 (Mdivi-1) during OGD preserves mitochondrial shape and motility and promotes axon function recovery. In contrast, preconditioning WM by applying Mdivi-1 only before OGD fails to conserve mitochondrial shape or motility and fails to benefit axon function. Our findings suggest that inhibition of mitochondrial fission during ischemia promotes axon function recovery, but is not sufficient to precondition WM against ischemia. These results raise caution in that approaches to preconditioning neuronal cell bodies may not successfully transform to functional improvement following ischemia.
Abstract
Mechanisms of ischemic preconditioning have been extensively studied in gray matter. However, an ischemic episode affects both the gray matter (GM) and white matter (WM) portions of the brain. Inhibition of mitochondrial fission is one of the mechanisms of preconditioning neuronal cell bodies against ischemia. Although axons are anatomical extensions of neuronal cell bodies, injury mechanisms differ between GM and WM. Indeed, axonal dysfunction is responsible for much of the disability associated with clinical deficits observed after stroke; however, the signaling process underlying preconditioning remains unexplored in axons. Using mouse optic nerve, which is a pure isolated WM tract, we show that mitochondria in myelinated axons undergo rapid and profuse fission during oxygen glucose deprivation (OGD) that is mediated by translocation of cytoplasmic Dynamin Related Protein-1 (Drp-1) to mitochondria. OGD-induced mitochondrial fission correlates with reduced mitochondrial motility and loss of axon function. Mitochondrial fragmentation and loss of motility become permanent during the recovery period. Inhibiting mitochondrial fission by administering mitochondrial division inhibitor-1 (Mdivi-1) during OGD preserves mitochondrial shape and motility and promotes axon function recovery. In contrast, preconditioning WM by applying Mdivi-1 only before OGD fails to conserve mitochondrial shape or motility and fails to benefit axon function. Our findings suggest that inhibition of mitochondrial fission during ischemia promotes axon function recovery, but is not sufficient to precondition WM against ischemia. These results raise caution in that approaches to preconditioning neuronal cell bodies may not successfully transform to functional improvement following ischemia.
Introduction
Ischemic preconditioning is an endogenous neuroprotective mechanism in which the brain protects itself against future injury by adapting to low doses of an insult (Cadet et al., 2009; Dirnagl et al., 2009). Clinical studies indicate that patients who suffer from a transient ischemic attack show improved clinical recovery after a stroke (Moncayo et al., 2000; Wegener et al., 2004). Therefore, elucidating the complex molecular mechanisms underlying preconditioning is a critical challenge in stroke research (Gidday, 2016). Recent work has shown that mitochondria actively participate in the preconditioning signaling pathway by generating reactive oxygen species (ROS) (Kowaltowski et al., 2000; McLeod et al., 2005; Hirata et al., 2011; Gundimeda et al., 2012) or by inhibiting mito-KATP channels (Auchampach et al., 1992; Yao and Gross, 1993; Jabůrek et al., 1998; Garlid et al., 2003). Mitochondria are dynamic organelles that undergo fission, fusion, and intracellular transport while conducting multiple physiological functions including the generation of ATP through oxidative phosphorylation, the buffering of cytosolic Ca2+, and the generation of ROS (van der Bliek et al., 2017). In normal healthy cells, fission and fusion are balanced to maintain mitochondria within a certain range of length appropriate for the maintenance of cellular physiology (Flippo and Strack, 2017). In response to injury, mitochondria undergo fragmentation into small dysfunctional units that in turn generate excessive amounts of ROS, causing cellular damage (Reddy et al., 2012; Balog et al., 2016; Golpich et al., 2017; Wu et al., 2017). Fission of mitochondria is mediated by Dynamin Reactive Protein-1 (Drp-1). Drp-1 is recruited to the mitochondrial outer membrane by a variety of adaptor proteins and then aggregates and forms oligomers in the shape of a ring at the site of future fission along mitochondria (Hatch et al., 2014; Flippo and Strack, 2017). Upon contraction of this ring formation, Drp-1 divides the mitochondrion into two separate mitochondria. Therefore, inhibition of Drp-1 activity has been of interest to attenuate or to prevent mitochondrial injury. In addition to genetic modifications, which produce serious effects on development and longevity (Ishihara et al., 2009; Wakabayashi et al., 2009), pharmacological blockade of Drp-1 with Mdivi-1, which is a blood-brain barrier-permeable inhibitor, provides a powerful tool with greater spatiotemporal control that can be acutely monitored and visualized.
A stroke affects both the gray matter (GM) and white matter (WM) portions of the brain (Mohr 2011). Faithful axon conduction is crucial for signaling among neuronal cell bodies to connect GM and WM in order to achieve and maintain proper function. Despite the anatomical perception that myelinated axons are a natural extension of neurons, axons are independent of their cell bodies in their supply of energy, metabolism, and injury mechanisms. It is important to note that approaches for neuronal protection fail to improve, or even impede, axon function recovery after ischemia (Tekkök et al., 2007; Baltan, 2009, 2012, 2014a). Inhibition of mitochondrial fission has been shown to induce preconditioning tolerance to neurons in vivo (Park et al., 2011b; Xie et al., 2013; Zhang et al., 2013a; ( Zuo et al., 2014, 2016; Jin et al., 2016; Kim et al., 2016; Deryagin et al., 2017) and in vitro (Correia et al., 2010; Wang et al., 2014), which raises the question as to whether or not preservation of mitochondria can precondition WM against ischemic injury.
Rodent brains contain only 10% WM by volume, as opposed to the human brain, which contains a significantly higher percentage (50% by volume) (Zhang and Sejnowski, 2000). Thus, the response to ischemia is dominated by neuronal injury in rodent brain. However, the use of optic nerve, which is a pure WM tract, avoids this impediment and provides a platform to test WM injury mechanisms in isolation. We hypothesized that blockade of mitochondrial fission during oxygen glucose deprivation (OGD) with Mdivi-1 will prevent mitochondrial fragmentation and preserve mitochondrial motility, thereby promoting axon function recovery and mediating ischemic tolerance in WM. We developed a novel technique for live imaging of axonal mitochondria to monitor mitochondrial fission and motility using optic nerves obtained from Thy-1 mito CFP (+) mice. Together with direct quantification of axon function, we aimed to reveal the interaction between mitochondrial dynamics and preconditioning in WM.
Materials and methods
Animals and Chemicals
All experimental procedures were approved by The Institutional Animal Care and Use Committee of the Cleveland Clinic. Mouse optic nerves (MONs) were obtained from C57BL/6J mice and from mice expressing mitochondrially-targeted cyan fluorescent protein (CFP) on a C57BL/6 background (Thy-1 mito-CFP (+); (Misgeld et al., 2007). Thy-1 mito-CFP (+) mice were originally obtained at the University of Washington and were later relocated to and bred at the Cleveland Clinic Foundation. Mitochondrial division inhibitor-1 (Mdivi-1) was purchased from Selleck Chemicals (#S7162, Houston, TX). The sources for other chemicals used have been described in detail previously (Baltan et al., 2008; Stahon et al., 2016).
Optic Nerve Preparation, Electrophysiological Recordings, and Oxygen Glucose Deprivation
Following CO2 asphyxiation, MONs were obtained from C57BL/6J or Thy-1 mito-CFP (+) mice at 2-3 months of age. MONs were gently cleared off from their dural sheaths and transferred to a Haas-Top perfusion chamber (Harvard Apparatus) and superfused with artificial cerebrospinal fluid (ACSF) containing the following (in mmol/L): 124 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 glucose. MONs were allowed to equilibrate for at least 15 min in the recording chamber in normal ACSF bubbled with a 95% O2/5% CO2 mixture. All electrophysiological recordings were performed at 37°C. Suction electrodes back-filled with glucose-free ACSF were used for stimulation and for recording compound action potentials (CAPs). The stimulation electrode was connected to a stimulus isolation unit (A365R Stimulus isolator; WPI, Sarasota, FL) and elicited CAPs at 30 s intervals. Stimulus pulse (30 μs duration) strength was adjusted to evoke the maximum CAP possible and then increased another 25% for supramaximal stimulation. The recording electrode was connected to an Axoclamp 900A amplifier (Molecular Devices, San Jose, CA) and the signal was amplified 20 or 50 times, filtered at 3 kHz (SR560, Stanford Research Systems, Sunnyvale, CA). OGD was induced as previously described (Baltan et al., 2008, 2010, 2011; Murphy et al., 2014; Stahon et al., 2016) by switching to glucose-free ACSF (replaced with equimolar sucrose to maintain osmolarity) and a gas mixture containing 95% N2/5% CO2. OGD was applied for 60 min, glucose-containing ACSF and O2 were restored, and CAPs were recorded for up to 5 h after the end of OGD. MONs from Thy-1 mito-CFP (+) mice were collected and fixed for subsequent CFP (+) mitochondrial analysis (see below).
Time-Lapse Live Imaging of Mitochondrial Fission and Motility
These experiments were performed exclusively on MONs obtained from Thy-1 mito-CFP (+) mice. After dissection, MONs were transferred to a C-Stim CMC Microscope Chamber System (IonOptix, Westwood, MA) attached to an in-line heater (Cell MicroControls MTCII temperature controller and heater; IonOptix, Westwood, MA) to keep the chamber at 37°C. The MONs were superfused with ACSF bubbled with a 95% O2/5% CO2 mixture at a flow rate of 3 ml/min. OGD was induced by switching to glucose-free ACSF saturated with 95% N2/5% CO2. To determine mitochondrial motility in both the anterograde and retrograde directions, Thy-1 mito-CFP (+) MONs were imaged every 1.5 s at 1024 x 1024 resolution from a single microscopic field using an inverted confocal microscope (Leica DMI6000, Buffalo Grove, IL, 40x water immersion objective, numerical aperture, 0.80). The imaging area was set to contain at least three axons (confirmed during analysis) in which there would be at least ten mitochondria moving in either direction. Time lapse images were captured during the last 5 min of baseline (20 min), during OGD (60 min), and during the recovery period (20 min). Imaging parameters were chosen and set to minimize photobleaching. Control experiments consisting of imaging MONs using the same parameters without OGD showed less than 10% CFP fluorescent loss over the same duration (data not shown). Mitochondrial motility, direction, and changes in mitochondrial size were analyzed using ImageJ software combined with the KymoTool Box plugin (Zala et al., 2013). Stationary and motile mitochondria, as well as the direction of motility, were identified and kymographs were generated in order to measure the anterograde and retrograde movements during baseline, OGD (60 min), and recovery.
Confocal Imaging and Pixel Intensity Analysis of Thy-1 mito-CFP (+) Mice
As previously described (Baltan et al., 2011; Baltan, 2012, 2014a; Murphy et al., 2014; Stahon et al., 2016), expression of CFP was imaged using a Leica DMI6000 inverted confocal laser-scanning microscope using Thy-1 mito-CFP (+) mice. Two to three adjacent sections for each MON were imaged with the wavelength set at 456 nm. A total of 10 optical sections of 1 μm thickness at 512 × 512 pixel size were collected in the z-axis from a single microscopic field using the 40x objective lens (HCX APO, water immersion; numerical aperture, 0.80) under fixed gain, laser power, pinhole, and photomultiplier tube settings. To compare pixel intensity, all sections were processed concurrently with Leica imaging software (LAS-AF version 2.7) using a single channel. The z-stacks were projected into a single plane image before analysis and assessment of pixel intensity.
Western blots
Mitochondria-rich lysate fractions and cytosol fractions were prepared from MON samples (two or three pairs; modified from (Disatnik et al., 2013). MON samples were homogenized in 75 µL of isolation buffer (300 mM sucrose, 10 mM HEPES, 2 mM EGTA, pH 7.2, at 4°C) with protease inhibitor and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO). After centrifugation at 950 g for 10 min, the supernatant was collected and further spun at 10,000 g for 20 minutes at 4°C. The final pellets suspended in lysis buffer were used as the mitochondrial-rich lysate fractions and the supernatants were used as cytosol fractions. Protein concentration was estimated using a BCA protein assay kit (Thermo Scientific, Rockford, IL). Protein lysates were prepared in 4x Laemmli sample buffer (Bio-Rad, Hercules, CA) and 2-mercaptoethanol at a ratio of 10:1 and then incubated at 95ºC for 10 minutes. Equal amounts of protein were loaded into each well (10-22 µg) of stain-free 4-20% Mini-Protean TGX gels (Bio-Rad, Hercules, CA). After gel electrophoresis, protein samples were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). Blots were blocked in 5% dry milk in TBS-Tween (TBS-T; 0.1%) for one hour at room temperature. Both primary (mouse Drp-1 #611112, BD Biosciences) and secondary antibodies were made in blocking buffer. Primary antibody was incubated overnight at 4ºC at 1:500. After three washes in TBS-T for 5 min, blots were incubated in secondary antibodies (goat anti-mouse HRP, Jackson Immunoresearch) at room temperature for 2 hours. Chemiluminescence was detected using Clarity Western ECL substrate (Bio-Rad, Hercules, CA), imaged using a Biorad Chemidoc MP, and analyzed using Image Lab software version 4.1 (Bio-Rad, Hercules, CA) for volumetric analysis of protein expression. Drp-1 levels were normalized to voltage-dependent anion channel (VDAC, # 10866-1-AP, Protein Tech Group Inc.) antibody levels in mitochondria-rich fractions and to b-actin (A5441, Sigma-Aldrich) in cytosol fractions, respectively.
Data Acquisition and Statistical Analysis
Optic nerve function was monitored quantitatively as the area under the supramaximal CAP using Clampfit (version 10.2, Molecular Devices, CA). CAP area is a complex spatiotemporal summation of action potentials from individual axons (Stys et al., 1991; Baltan et al., 2008, 2011; Stahon et al., 2016). For comparing CAP area among experimental groups, data from each group of experiments were pooled and CAP areas were normalized to the baseline conditions. Data were normalized by setting the mean of initial baseline values (measured over 15 min) to a value of 1 as previously described (Baltan et al., 2008, 2011; Stahon et al., 2016). All data are presented as mean ± SEM. Graphpad Prism (ver. 4.0c, La Jolla, CA) was used for statistical analysis. For sample sets containing more than two groups, one-way ANOVA followed by post hoc Bonferroni’s test for between-group comparisons was used. The p values and significance values are indicated individually for each figure in the text. The n values indicate the number of optic nerves and the numbers in brackets denote mitochondria followed by experiment numbers in histograms quantifying mitochondrial motility.
Results
Drp-1 regulates ischemia-induced mitochondrial fission
Mitochondrial length is determined by the balance between the rates of mitochondrial fission and fusion and is important for controlling the spatiotemporal properties of mitochondrial responses during physiological and pathophysiological processes (Szabadkai and Duchen, 2008). Because ischemia caused extensive fission and fragmentation, leading to smaller-sized mitochondria that correlated with irreversible axon function loss (Baltan et al., 2011), we reasoned that preventing mitochondrial fission could maintain mitochondrial size, shape, and morphology, and also precondition WM as characterized by improved axon function recovery. Drp-1, which is a mitochondrial fission protein, is critical for mitochondrial division, size, and shape (Reddy et al., 2011) and is regulated with age in WM (Stahon et al., 2016). Mdivi-1 is a selective inhibitor of Drp-1 and has been shown to confer protective effects in heart, kidney, retinal ganglion cells, spinal cord, and cerebral ischemia reperfusion models (Brooks et al., 2009; Ong et al., 2010; Park et al., 2011a; Grohm et al., 2012; Liu et al., 2015). Mdivi-1 is reported to suppress the translocation of cytosolic Drp-1 onto mitochondria to prevent fission (Kim et al., 2017; Valenti et al., 2017). To ensure that Mdivi-1 acted on Drp-1 and conserved mitochondrial size in myelinated axons of optic nerve, we quantified the protein levels of Drp-1 in the cytosolic (Figure 1A and B, left panels) and mitochondrial (Figure 1A and B, right panels) fractions. OGD suppressed Drp-1 in the cytosolic fraction (0.7 ± 0.04%, n = 4) because it is translocated to the mitochondrial fraction (2.0 ± 0.4%, n = 6; control 1.0 ± 0.0, n = 7, p < 0.05, one-way ANOVA, Bonferroni’s post hoc test), while Mdivi-1 (50 µM) application reversed these effects of OGD. Cytosolic Drp-1 levels increased to 1.55 ± 0.3%, (n = 3, p < 0.05, one-way ANOVA, Bonferroni’s post hoc test) and mitochondrial Drp-1 levels dropped to 0.94 ± 0.2, (n = 6, p < 0.05, one-way ANOVA, Bonferroni’s post hoc test) in MONs pre-treated with Mdivi-1. Consequently, Mdivi-1 application preserved mitochondrial size and CFP (+) fluorescence (Figure 1C, Mdivi-1 throughout). In control optic nerves obtained from Thy-1 mito-CFP (+) mice, axonal mitochondria displayed elongated tubular CFP (+) structures (Figure 1C, Control). After OGD, as reported previously (Baltan et al., 2011), there was a dramatic reduction in CFP fluorescence and remaining mitochondria presented a punctuate morphology (Figure 1C, OGD) consistent with ischemia-induced fission. Pretreatment of MONs with Mdivi-1 preserved CFP pixel intensity and mitochondrial morphology (Figure 1C, Mdivi-1 throughout), confirming the principal mode of action of Mdivi-1 inhibition of mitochondrial fission in myelinated axons of optic nerve and that diffuse mitochondrial fission observed during OGD irreversibly hampers axon function recovery.
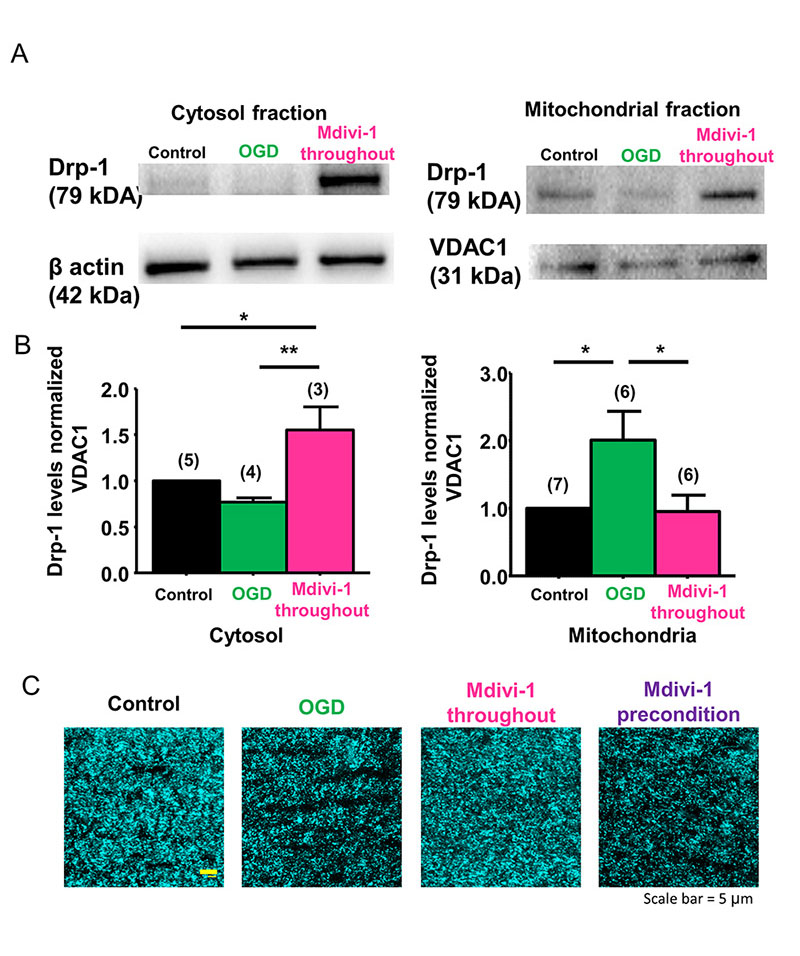
In a new window | Download PPT
Figure 1: Drp-1 regulates ischemia-induced mitochondrial fission in WM. (A, B) OGD suppressed Drp-1 levels in MONs in the cytosolic fraction (0.7 ± 0.04%, n = 4) and revealed a corresponding increase in the mitochondrial fraction (2.0 ± 0.4%, n = 6; control 1.0 ± 0.0, n = 7, p< 0.05). Following application of the Drp-1 inhibitor Mdivi-1 (50 µM), cytosolic Drp-1 levels increased to 1.55 ± 0.3%, (n = 3, p < 0.05) and mitochondrial Drp-1 levels dropped to 0.94 ± 0.2% (n = 6, p < 0.05). (C) Confocal microscopy imaging showed that OGD drastically reduced CFP fluorescence in MONs from Thy-1 mito CFP (+) mice. Note that mitochondrial numbers declined and mitochondria became smaller and more round with OGD. Mdivi-1 application before, during, and after OGD preserved CFP pixel intensity and mitochondrial morphology; however, Mdivi-1 preconditioning application failed to preserve CFP fluorescence. (Scale bar, 5 µm). n = number of MONs; *p < 0.05, **p < 0.01, one-way ANOVA. Error bars indicate SEM.
Inhibition of mitochondrial fission promotes myelinated axon function recovery following ischemia
To assess whether inhibition of mitochondrial fission promoted axon function recovery, the effects of OGD on CAP area recovery with or without Mdivi-1 was tested using different concentrations of Mdivi-1 (Figure 2A). The functional integrity of axons was quantified by the area under the evoked CAPs. After a 30-min baseline recording, Mdivi-1 was introduced at 1, 10, 20, or 50 µM concentrations (pink) for 30 min and the superfusion conditions were maintained for 60 min of OGD and the initial 30 min of reperfusion time (Figure 2A and C). After OGD, CAP area recovered to 21.6 ± 1.8% (n = 14, green) of the maximum-recorded CAP area, whereas a lower dose of Mdivi-1 (1 µM) improved CAP area recovery to 25.9 ± 6.4% (n = 2, p < 0.4) and higher doses (10, 20, and 50 µM) improved CAP area recovery to 41.8 ± 4.1% (n = 2 p < 0.02), 37.4 ± 23.5% (n = 2, p < 0.4), and 46.4 ± 10.3% (n = 4, p < 0.003, one-way ANOVA, Bonferonni’s post hoc test), respectively (Figure 2A). Application of 50 µM Mdivi-1 provided the greatest amount of protection (Figure 2A-C, pink) when applied throughout the procedure (30 min before, 60 min during, and 30 min after the end of OGD; Figure 2C, pink time course and inset histograms). As expected, this functional recovery correlated with inhibition of mitochondrial fission and consequent preservation of mitochondrial integrity (Figure 1C, Mdivi-1 throughout).
Inhibition of mitochondrial fission fails to precondition myelinated axons against ischemia
Mitochondria have been implicated in neuroprotective signaling underlying preconditioning and represent a promising target in various neurodegenerative diseases (Correia et al., 2010). It was proposed that low levels of mitochondrial ROS generation induces fission and fusion of mitochondria and alters mitochondrial motility and dynamics to form a mitochondria-free gap, thereby preventing propagation of ROS during oxidative injury to precondition the tissue (Jou, 2008; Correia et al., 2010). We reasoned that by inhibiting mitochondrial fission, Mdivi-1 can regulate mitochondrial motility and subsequently precondition WM to promote axon function recovery as previously reported in GM (Ravati et al., 2000, 2001; Dirnagl and Meisel, 2008; Jou, 2008; Dirnagl et al., 2009). To determine whether inhibition of mitochondrial fission was effective in promoting axon function recovery when applied before the onset of OGD, Mdivi-1 was applied for 30 min after obtaining a stable baseline and then OGD was induced in normal ACSF (Figure 2B and C, purple traces and time course). OGD predictably suppressed the CAP area completely (Figure 2B, purple traces), and preconditioning with Mdivi-1 (50 µM) failed to promote axon function recovery (Figure 2B and C, purple time course and inset). Consistent with these results, Mdivi-1 application before OGD failed to prevent mitochondrial fission or to preserve mitochondrial size and shape (Figure 1C, right panel).
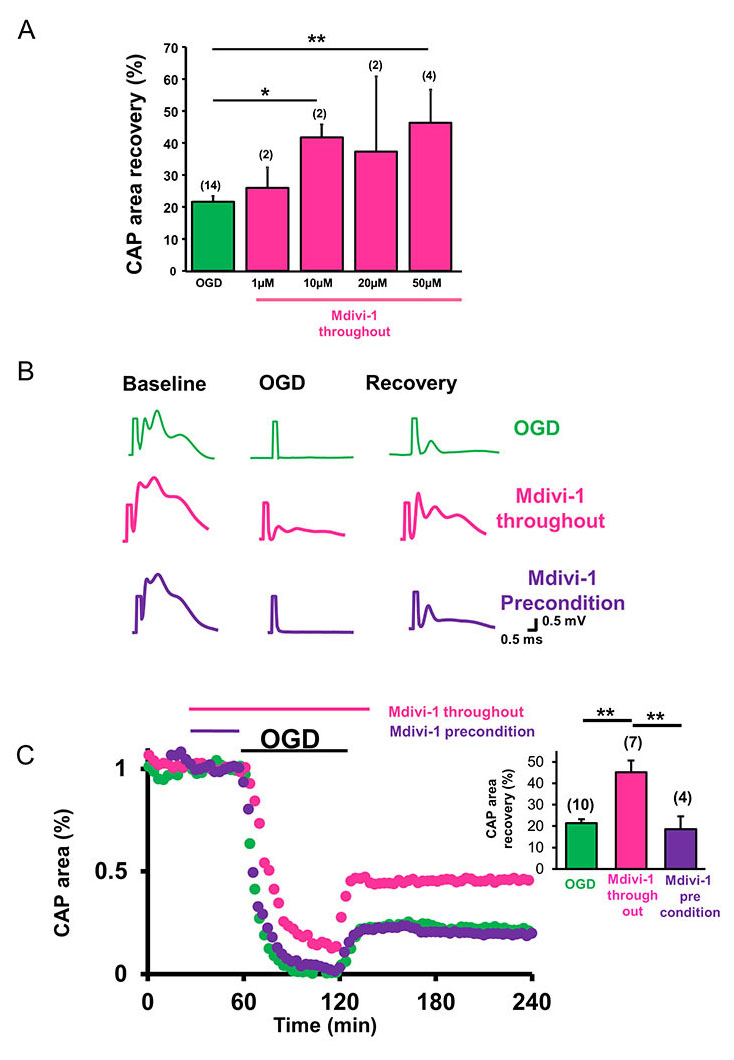
In a new window | Download PPT
Figure 2: Inhibition of mitochondrial fission promotes WM function after ischemia. (A) Mdivi-1 exerts dose-dependent protection of axon function recovery in MONs following OGD. (B) Representative CAP traces at baseline in control ACSF (a), 60 min OGD (b), and recovery (c) conditions are shown for control (green), Mdivi-1 applied as an all-through treatment (pink), and Mdivi-1 applied as a preconditioning treatment (purple). (C) CAP area recovered minimally after OGD (green, 21.6 ± 1.8%, n = 10). Mdivi-1 all-through application (pink) preserved CAP area during OGD and improved CAP area recovery during reperfusion (41 ± 5.0%, n = 7). Mdivi-1 preconditioning treatment (purple) failed to promote axon function recovery (18.6 ± 6.0%, n = 4). Pink and purple horizontal lines represent Mdivi-1 all-through and preconditioning treatment applications, respectively. The inset histograms show quantification of CAP area recovery 2 h after OGD. MONs pretreated with Mdivi-1 (pink) showed improved axon function recovery, whereas the CAP area recovery failed to improve when MONs were preconditioned with Mdivi-1 (purple). n = number of MONs; *p < 0.05, **p < 0.01, one-way ANOVA. Error bars indicate SEM.
Mitochondrial fission alters mitochondrial motility
Mitochondria underwent extensive fission during ischemia, but whether mitochondrial fragmentation correlates with loss of motility and whether preservation of mitochondrial motility correlates with axon function recovery were still unanswered questions. Time-lapse live imaging of mitochondria showed that mitochondria move bi-directionally, change direction, or become stationary in response to OGD (Figure 3). Optic nerves were placed and stabilized for 15 min in a perfusion chamber and continuously superfused with oxygenated ACSF (95% O2/5% CO2) at a rate of 3 ml/min at 37°C. Once an area was chosen where mitochondria in myelinated axons were clearly visible, mitochondria were imaged every 1.5 s. These mitochondria were observed to be very dynamic organelles that underwent fission and fusion constantly. Coordinated motility of mitochondria ensures that metabolically active areas are adequately supplied with ATP such that injured mitochondria are replaced with healthy ones during injury. Motile and stationary mitochondria were identified using kymographs (Figure 3A), which represent 2-dimensional images of stationary and motile mitochondria during the entire time-lapse sequence. Vertical lines represent the stationary mitochondria (Figure 3A), while oblique lines represent motile mitochondria (Figure 3A). Specifically, blue lines denote mitochondria moving in the anterograde direction and tan lines indicate mitochondria moving in the retrograde direction. Note that a small but similar number of mitochondria moved in either direction (Figure 3A, tan and blue lines), while the majority remained stationary (Figure 3A, vertical lines). The size of mitochondria in young myelinated axons varies between 1-8 µm and the average mitochondrial velocity varies between 0.6 - 1.7 µm/s (Stahon et al., 2016). Mitochondria maintained a stable speed under control conditions. Onset of OGD caused a 50% drop in mitochondrial motility both in the anterograde (Figure 3B and 3C, blue histograms, OGD 0.57 ± 0.03, n = 157 vs. Baseline 1.0 ± 0.02, n = 318, p < 0.001, one-way ANOVA, Bonferonni’s post hoc test) and retrograde directions (Figure 3B and 3C, tan histograms, OGD 0.48 ± 0.03, n = 95 vs. Baseline 1.0 ± 0.03, n = 243, p < 0.001, one-way ANOVA, Bonferonni’s post hoc test). Recovery in the anterograde direction was not different (Figure 3B and 3C, blue histograms, Recovery 0.71 ± 0.04, n = 139, p > 0.05, one-way ANOVA, Bonferonni’s post hoc test). Interestingly recovery in the retrograde direction was improved (Figure 3B and 3C, tan histograms, Recovery 0.73 ± 0.03, n = 162, p < 0.001, one-way ANOVA, Bonferonni’s post hoc test). Mdivi-1 caused a prominent increase in mitochondrial motility in both directions within 10 min of application (Figure 3B, pink histograms, one-way ANOVA, p < 0.001), suggesting that inhibition of mitochondrial fission enhanced mitochondrial transport within myelinated axons. Note that the baseline effect of Mdivi-1 was more prominent in the anterograde direction (Figure 3B Anterograde, pink histograms with blue border, Mdivi-1 throughout Baseline 1.60 ± 0.09, n = 68, p < 0.001) when compared to the retrograde direction (Figure 3B Retrograde, pink histograms with tan border, Mdivi-1 throughout Baseline 1.39 ± 0.08, n = 63, p < 0.001, one-way ANOVA, Bonferonni’s Post-test). This increase in mitochondrial motility persisted during OGD (60 min) (Figure 3B Anterograde, pink histograms with blue border, Mdivi-1 throughout OGD 0.89 ± 0.06, n = 53, vs. OGD 0.57 ± 0.03, n = 157, p < 0.001; Retrograde, pink histograms with tan border, Mdivi-1 throughout OGD 0.87 ± 0.04, n = 69, vs. OGD 0.48 ± 0.03, n = 95, p < 0.001, one-way ANOVA, Bonferonni’s post hoc test) and recovery (20 min) (Figure 3B Anterograde, pink histograms with blue border, Mdivi-1 throughout Recovery 0.99 ± 0.03, n = 72, vs. Recovery 0.71 ± 0.04, n = 139, p < 0.001; Retrograde, pink histograms with tan border, Mdivi-1 throughout Recovery 0.95 ± 0.05, n = 55, vs. Recovery 0.73 ± 0.03, n = 162, p < 0.05, one-way ANOVA, Bonferonni’s post hoc test) in both directions. Together with the electrophysiology results, these findings suggest that application of Mdivi-1 promotes axon function recovery and preserves mitochondrial motility by preventing mitochondrial fission during ischemia.

In a new window | Download PPT
Figure 3: Mdivi-1 preserves mitochondrial motility against ischemia only when applied during OGD. (A) A representative kymograph created from CFP (+) live mitochondrial imaging shows immotile mitochondria as vertical lines. Note the diagonal lines representing mitochondrial movement in axons in the anterograde direction (blue) as well as the retrograde direction (tan). (B) Quantification of mitochondrial motility from kymographs demonstrated a 50% reduction in mitochondrial motility, both in the anterograde (blue histograms) and retrograde directions (tan histograms), which showed minimal recovery in the anterograde direction reperfusion. Mdivi-1 caused a prominent increase in mitochondrial motility in both directions within 10 min of application (pink histograms), suggesting that inhibition of mitochondrial fission enhanced mitochondrial transport within the myelinated axons. Note that the baseline effect of Mdivi-1 was more prominent in the anterograde direction (pink with blue border histograms). This increase in mitochondrial motility persisted during OGD (60 min) and recovery (20 min) in both directions. n = number of MONs; *p < 0.05, *** p < 0.001, one-way ANOVA with Bonferonni’s post hoc test. (C) Mitochondrial motility analysis showed a similar increase in mitochondrial motility in both directions upon preconditioning application of Mdivi-1 (purple histograms). However, mitochondrial motility was suppressed during OGD and did not improve during the recovery period in either the anterograde or retrograde direction. *** p < 0.001, one-way ANOVA with Bonferonni’s post hoc test.
To confirm that preservation of mitochondrial motility predicts the extent of axon function recovery, we applied Mdivi-1 just before OGD for 30 min. Live-imaging of mitochondria showed a similar increase in mitochondrial motility in both directions upon application of Mdivi-1 (Figure 3C Anterograde, purple histograms with blue border, Mdivi-1 throughout Baseline 1.60 ± 0.09, n = 68, vs. Baseline 1.0 ± 0.02, n = 318, p < 0.001; Figure 3B Retrograde, purple histograms with tan border, Mdivi-1 throughout Baseline 1.39 ± 0.08, n = 63, vs. Baseline 1.0 ± 0.03, n = 243, p < 0.001, one-way ANOVA, Bonferonni’s post hoc test). However, mitochondrial motility was suppressed during OGD (Figure 3C Anterograde, purple histograms with blue border, Mdivi-1 throughout OGD 0.74 ± 0.03, n = 62, vs. OGD 0.57 ± 0.03, n = 157, p > 0.05; Retrograde, purple histograms with tan border, Mdivi-1 throughout OGD 0.66 ± 0.04, n = 53, vs. OGD 0.48 ± 0.03, n = 95, p > 0.05, one-way ANOVA, Bonferonni’s post hoc test) and did not improve during the recovery period (Figure 3C Anterograde, purple histograms with blue border, Mdivi-1 throughout Recovery 0.76 ± 0.05, n = 42, vs. Recovery 0.71 ± 0.04, n = 139, p > 0.05; Retrograde, purple histograms with tan border, Mdivi-1 throughout Recovery 0.72 ± 0.05, n = 38, vs. Recovery 0.73 ± 0.03, n = 162, p > 0.05, one-way ANOVA, Bonferonni’s post hoc test), which is consistent with failure of axon function recovery. Therefore, we propose that inhibition of mitochondrial fission directs mitochondrial motility, which consequently determines axon function recovery, and that preservation of mitochondrial motility is an indication of improved axon function recovery.
Discussion
Our results showed that inhibition of mitochondrial fission, specifically inhibition of Drp-1 by Mdivi-1, during an ischemic episode modifies mitochondrial motility and determines the extent of axon function recovery in a completely myelinated WM tract, the optic nerve. This protective effect of Mdivi-1 was dose-dependent and correlated with preservation of mitochondrial integrity. While these results propose mitochondrial dynamics as a plausible target to reduce ischemic injury and to restore function in WM, Drp-1 inhibition failed to offer ischemic preconditioning tolerance to myelinated axons, in contrast to its effect on neuronal cell bodies (Grohm et al., 2012; Zhang et al., 2013a; Wang et al., 2014; Cui et al., 2016). We propose that regulating mitochondrial dynamics is necessary to promote functional recovery, but is not sufficient to provide ischemic tolerance to WM function.
One of the main findings of our study is that inhibition of Drp-1 during ischemia prevents mitochondrial fission, preserves CFP fluorescence, sustains axon conduction, and confers a dose-dependent promotion of axon function recovery. In myelinated axons, an ischemic episode causes widespread mitochondrial fission mediated by translocation of cytoplasmic Drp-1 onto mitochondria. This in turn reduces the ability of mitochondria to produce the ATP that is essential for sustaining the excitability and function of axons (Baltan et al., 2011; Baltan, 2012, 2014b). Consequently, axon function is completely lost during injury (Tekkök et al., 2007; Baltan et al., 2008, 2013; Baltan, 2014a). During recovery, mitochondrial fission became permanent, as characterized by the loss of CFP (+) fluorescence in Thy-1 mito-CFP (+) mice and subsequently a small portion of axons (~20%) regained function (Tekkök et al., 2007; Baltan et al., 2008, 2013; Baltan, 2014a). The reported beneficial effects on neurons and improved functional recovery in our study make Mdivi-1 an ideal candidate for therapy to restore function when applied during ischemia.
A novel finding in our study was that Mdivi-1 not only prevents mitochondrial fission, but also enhances mitochondrial motility without affecting normal axon function. Mdivi-1 application preserved mitochondrial motility in both the anterograde and retrograde directions during OGD and during recovery, which was associated with improved axon function recovery. This suggests that mechanisms of fission and mitochondrial motility are interconnected. Previous studies have showed that Miro, which is a Ca2+-sensing member of the microtubular mitochondrial complex that determines the motility of mitochondria, depending upon the availability of ATP and Ca2+, is intricately linked to the mitochondrial fission protein Drp-1 (Saotome et al., 2008). Miro adaptor protein exhibits GTPase activity and forms complexes with kinesin for anterograde mitochondrial transport (Guo et al., 2005; Russo et al., 2009) and dynein for retrograde mitochondrial transport (Russo et al., 2009; Morlino et al., 2014; Melkov et al., 2016) along microtubules. Mdivi-1 preserves ATP levels during ischemia reperfusion injury (Li et al., 2016), thus effectively abolishing impairments to mitochondrial movement in both directions and blocking mitochondrial fission to improve axon function recovery.
An unexpected aspect of our study was the lack of preconditioning-induced tolerance when Mdivi-1 was applied only before injury. Many lines of evidence have posited that mitochondria constitute a convergence point of preconditioning in in vitro and in vivo models of cerebral ischemia (Perez-Pinzon et al., 2005; Dave et al., 2006). Mdivi-1 is a small molecule that is readily permeant through the blood-brain barrier and it provides ischemic tolerance to neurons by maintaining mitochondrial integrity and function. Previously, we reported that maintaining mitochondrial integrity during or after ischemia provides axon function protection and promotes recovery by conserving ATP production and reducing excitotoxicity (Baltan et al., 2011, 2013; Baltan, 2014b). Among well-established mediators of preconditioning (Barone et al., 1998; Kowaltowski et al., 2000; McLeod et al., 2005; Dave et al., 2006; Cadet et al., 2009; Dirnagl et al., 2009; Marsh et al., 2009; Hirata et al., 2011; Gundimeda et al., 2012; Hibert et al., 2013; Hamner et al., 2015), recent studies link mitochondrial biogenesis, specifically dynamics to preconditioning (Correia et al., 2010). Mdivi-1 application led to a prominent increase in mitochondrial motility during baseline conditions. When Mdivi-1 was applied during OGD, mitochondrial motility was conserved during ischemia and improved further during recovery. This increase in mitochondrial motility, together with preserved mitochondrial integrity, implicated Mdivi-1 application as an ideal candidate to induce ischemic tolerance to WM against injury, comparable to the protection achieved in neurons. However, applying Mdivi-1 only before injury failed to precondition WM against ischemia. This is not due to a general lack of Mdivi-1 effects when applied only before ischemia, since our live imaging results confirm that Mdivi-1 application instantly modifies mitochondrial dynamics, as evidenced by their enhanced motility during the baseline period. This increase in motility is preserved during OGD, albeit to a smaller extent compared to Mdivi-1 application during ischemia. However, the enhanced motility of mitochondria vanishes during the recovery period. Based on the interplay between mitochondrial motility and mitochondrial fission, it is plausible that motile mitochondria are an indicator of better functional recovery. In particular, the extent of mitochondrial motility during recovery directs functional recovery. The downstream molecular mechanisms of this protection are currently under investigation.
In conclusion, this is the first study to investigate whether inhibition of mitochondrial fission induces ischemic preconditioning tolerance to axons in an isolated pure WM tract. Conserving mitochondrial structure sustains mitochondrial motility during ischemia and confers functional protection. However, this approach fails to precondition axon function against ischemia, in contrast to its protective effects on neuronal survival. These findings support the concept that manipulations to precondition the brain should consider interventions to be beneficial for both the gray and white matter.
Acknowledgements
The authors thank Dr. Chris Nelson for his assistance in editing the manuscript. These studies were funded by NIH grants R56AG033720 and R01AG033720 to S.B.
References
Chinthasagar Bastian
1Department of Neurosciences, Cleveland Clinic Foundation, Cleveland, Ohio 44195
Stephen Politano
1Department of Neurosciences, Cleveland Clinic Foundation, Cleveland, Ohio 44195
Jerica Day
1Department of Neurosciences, Cleveland Clinic Foundation, Cleveland, Ohio 44195
Andrew McCray
1Department of Neurosciences, Cleveland Clinic Foundation, Cleveland, Ohio 44195
Sylvain Brunet
1Department of Neurosciences, Cleveland Clinic Foundation, Cleveland, Ohio 44195
Selva Baltan
Department of Neurosciences, Cleveland Clinic Foundation, Cleveland, Ohio 44195
Corresponding author
Selva Baltan
Email: baltans@ccf.org
In a new window | Download PPT
Figure 1: Drp-1 regulates ischemia-induced mitochondrial fission in WM. (A, B) OGD suppressed Drp-1 levels in MONs in the cytosolic fraction (0.7 ± 0.04%, n = 4) and revealed a corresponding increase in the mitochondrial fraction (2.0 ± 0.4%, n = 6; control 1.0 ± 0.0, n = 7, p< 0.05). Following application of the Drp-1 inhibitor Mdivi-1 (50 µM), cytosolic Drp-1 levels increased to 1.55 ± 0.3%, (n = 3, p < 0.05) and mitochondrial Drp-1 levels dropped to 0.94 ± 0.2% (n = 6, p < 0.05). (C) Confocal microscopy imaging showed that OGD drastically reduced CFP fluorescence in MONs from Thy-1 mito CFP (+) mice. Note that mitochondrial numbers declined and mitochondria became smaller and more round with OGD. Mdivi-1 application before, during, and after OGD preserved CFP pixel intensity and mitochondrial morphology; however, Mdivi-1 preconditioning application failed to preserve CFP fluorescence. (Scale bar, 5 µm). n = number of MONs; *p < 0.05, **p < 0.01, one-way ANOVA. Error bars indicate SEM.
In a new window | Download PPT
Figure 2: Inhibition of mitochondrial fission promotes WM function after ischemia. ) A(Mdivi-1 exerts dose-dependent protection of axon function recovery in MONs following OGD. (B) Representative CAP traces at baseline in control ACSF (a), 60 min OGD (b), and recovery (c) conditions are shown for control (green), Mdivi-1 applied as an all-through treatment (pink), and Mdivi-1 applied as a preconditioning treatment (purple). (C) CAP area recovered minimally after OGD (green, 21.6 ± 1.8%, n = 10). Mdivi-1 all-through application (pink) preserved CAP area during OGD and improved CAP area recovery during reperfusion (41 ± 5.0%, n = 7). Mdivi-1 preconditioning treatment (purple) failed to promote axon function recovery (18.6 ± 6.0%, n = 4). Pink and purple horizontal lines represent Mdivi-1 all-through and preconditioning treatment applications, respectively. The inset histograms show quantification of CAP area recovery 2 h after OGD. MONs pretreated with Mdivi-1 (pink) showed improved axon function recovery, whereas the CAP area recovery failed to improve when MONs were preconditioned with Mdivi-1 (purple). n = number of MONs; *p < 0.05, **p < 0.01, one-way ANOVA. Error bars indicate SEM.
In a new window | Download PPT
Figure 3: Mdivi-1 preserves mitochondrial motility against ischemia only when applied during OGD.) Quantification of mitochondrial motility from kymographs demonstrated a 50% reduction in mitochondrial motility, both in the anterograde (blue histograms) and retrograde directions (tan histograms), which showed minimal recovery in the anterograde direction reperfusion. B) A representative kymograph created from CFP (+) live mitochondrial imaging shows immotile mitochondria as vertical lines. Note the diagonal lines representing mitochondrial movement in axons in the anterograde direction (blue) as well as the retrograde direction (tan). (A (Mdivi-1 caused a prominent increase in mitochondrial motility in both directions within 10 min of application (pink histograms), suggesting that inhibition of mitochondrial fission enhanced mitochondrial transport within the myelinated axons. Note that the baseline effect of Mdivi-1 was more prominent in the anterograde direction (pink with blue border histograms). This increase in mitochondrial motility persisted during OGD (60 min) and recovery (20 min) in both directions. n = number of MONs; *p < 0.05, *** p < 0.001, one-way ANOVA with Bonferonni’s post hoc test. (C) Mitochondrial motility analysis showed a similar increase in mitochondrial motility in both directions upon preconditioning application of Mdivi-1 (purple histograms). However, mitochondrial motility was suppressed during OGD and did not improve during the recovery period in either the anterograde or retrograde direction. *** p < 0.001, one-way ANOVA with Bonferonni’s post hoc test.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 8246 | 44 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA