Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Thrombin-induced tolerance against oxygen-glucose deprivation in astrocytes: role of protease-activated receptor-1
Time:2018-02-17
Number:11220
Author Affiliations

Conditioning Medicine, 2018. 1(2): 57-63.
Abstract
Our previous studies have found that pretreatment with a low dose of thrombin (thrombin preconditioning, TPC) reduces infarct volume and attenuates brain edema after focal cerebral ischemia in vivo and protects against the neuronal death induced by oxygen glucose deprivation (OGD) in vitro. In this study, we found that TPC (24 hours exposure to 0.5 or 1 U/ml thrombin) protects against OGD-induced astrocyte death, and that such protection is through protease-activated receptor-1 (PAR-1) and the p44/42 mitogen-activated protein kinase (MAPK)/p90 ribosomal S6 kinase (p90RSK) /heat shock protein 25 (HSP25) pathway. In contrast, in PAR-1 KO mouse astrocytes, TPC had no protective effect and it did not significantly phosphorylate p44/42 MAPK or p90RSK, or upregulate HSP25. PD98059, an inhibitor of p44/42 MAPK, blocked thrombin-induced tolerance as well as upregulation of phosphorylated p90RSK and HSP25 in WT mouse astrocytes. Furthermore, SL0101, an inhibitor of p90RSK, blocked thrombin-induced protection and the HSP25 upregulation in WT mouse astrocytes. These results suggest that TPC-induced tolerance in ischemic astrocytes may be through activation of thrombin receptor PAR-1 and a downstream p44/42 MAPK/p90RSK/HSP25 pathway.
Key words: Thrombin preconditioning, protease activated receptor-1, p44/42 MAPK, p90 ribosomal S6 kinase, heat shock protein 25
Abstract
Our previous studies have found that pretreatment with a low dose of thrombin (thrombin preconditioning, TPC) reduces infarct volume and attenuates brain edema after focal cerebral ischemia in vivo and protects against the neuronal death induced by oxygen glucose deprivation (OGD) in vitro. In this study, we found that TPC (24 hours exposure to 0.5 or 1 U/ml thrombin) protects against OGD-induced astrocyte death, and that such protection is through protease-activated receptor-1 (PAR-1) and the p44/42 mitogen-activated protein kinase (MAPK)/p90 ribosomal S6 kinase (p90RSK) /heat shock protein 25 (HSP25) pathway. In contrast, in PAR-1 KO mouse astrocytes, TPC had no protective effect and it did not significantly phosphorylate p44/42 MAPK or p90RSK, or upregulate HSP25. PD98059, an inhibitor of p44/42 MAPK, blocked thrombin-induced tolerance as well as upregulation of phosphorylated p90RSK and HSP25 in WT mouse astrocytes. Furthermore, SL0101, an inhibitor of p90RSK, blocked thrombin-induced protection and the HSP25 upregulation in WT mouse astrocytes. These results suggest that TPC-induced tolerance in ischemic astrocytes may be through activation of thrombin receptor PAR-1 and a downstream p44/42 MAPK/p90RSK/HSP25 pathway.
Key words: Thrombin preconditioning, protease activated receptor-1, p44/42 MAPK, p90 ribosomal S6 kinase, heat shock protein 25
Introduction
Thrombin, a serine protease, has an important role in brain injury and recovery after ischemic and hemorrhagic stroke. High concentrations of thrombin cause brain injury, but low concentrations exert neuroprotective effects (Hu et al., 2010; Masada et al., 2000; Striggow et al., 2000; Jiang et al., 2002; Hua et al., 2003; Choi et al., 2005; Henrich-Noack et al., 2006). Pretreatment with a low dose of thrombin attenuates the brain edema induced by thrombin, iron or brain hemorrhage; reduces infarct size in a rat middle cerebral artery occlusion model; and reduces brain damage in a rat model of Parkinson’s disease (Xi et al., 1999; Masada et al., 2000; Hua et al., 2003; Cannon et al., 2005; Cannon et al., 2006; Chen-Roetling et al., 2012; Guan et al., 2016). We have also found that prior treatment of a low dose of thrombin protects against the ischemic injury in rat neurons in vitro (Hu et al., 2010). This phenomenon has been termed as thrombin preconditioning (TPC), or thrombin-induced brain tolerance.
The precise mechanisms of thrombin-induced brain tolerance to ischemic and hemorrhagic stroke are not well known. However, activation of thrombin receptors and upregulation of mitogen-activated protein kinases, iron-handling proteins, aquaporin 4 and heat shock proteins in the brain may be associated with the induced tolerance (Hu et al., 2010; Xi et al., 1999; Xi et al., 2000; Hua et al., 2003; Yang et al., 2006; Hirt et al., 2009). Many previous studies have demonstrated that thrombin receptors are one of seven transmembrane G protein-coupled receptors that are activated by proteolytic cleavage rather than by ligand binding. Of the protease-activated receptor (PAR) family, three of them, PAR-1, PAR-3 and PAR-4, are activated by thrombin whereas PAR-2 is activated by trypsin and mast cell tryptase (Coughlin, 1999, 2000). PAR-1 expression is widely found in neurons, astrocytes, oligodendrocytes and microglia, and there is functional evidence for the presence of PAR-1 on all cell types (Weinstein et al., 1995; Gingrich and Traynelis, 2000; Ubl et al., 2000; Suo et al., 2003; Junge et al., 2004; Wang et al., 2006).
Preconditioning-induced neuroprotection may be associated with new protein synthesis (Gidday, 2016; Dirnagl et al., 2009). Activation of the p44/42 MAPK/p90RSK pathway plays an important role in protein synthesis leading to cell survival (Angenstein et al., 1998; Frodin and Gammeltoft, 1999; Anjum and Blenis, 2008). Heat shock protein 25 (HSP25, the murine HSP27), a small heat shock protein, can be markedly induced in astrocytes, and it has cytoprotective effects in a range of disease models, including cardiac ischemia, kainate-induced hippocampal cell death, nerve injury and cerebral ischemia (van der Weerd et al., 2010; Stetler et al., 2009; Shi et al., 2017).
In the present study, we investigated the effects of TPC on oxygen glucose deprivation (OGD)-induced death and the role of the PAR-1/p44/42 MAPK/p90RSK/HSP25 pathway in thrombin-induced neuroprotection in vitro using mouse primary astrocyte cultures.
Methods
Primary astrocytic culture
Primary astrocytic cultures were prepared from the brains of 1-3-day-old WT C57BL/6J mice (Jackson Laboratory) or PAR-1 KO mice (Jackson Laboratory) as described previously (He et al., 2010) with some modifications. Briefly, cerebral cortex was isolated, meninges and blood vessels were removed, and the tissue was dissociated by trypsinization and plated on 75 mm2 flasks in glial medium (Dulbecco’s modified Eagle medium with 10% horse serum, 10% fetal calf serum, 0.5 mM glutamine and 2% Antibiotic-Antimycotic; Gibco). The cells were kept at 37°C with 5% CO2, and growth medium was changed every other day. Twelve to 14 days after plating, the flasks were shaken at 200 rpm on a gyratory shaker for 1.5 hours at 4°C. After shaking, the flask-adherent cells were predominantly astrocytes. The supernatant was removed and the adherent cells were detached and plated on poly-L-lysine-coated 24-well plates (105 cells/well), 10-cm dishes (5 × 106 cells/dish) and 4-well chamber slides (5 × 104 cells/well).
Experimental groups
There were 3 parts to this study:
Part I. Experiments were performed to find whether the neuroprotective effect of TPC was through PAR-1 and its downstream kinases and protein. First, astrocytes from WT or PAR-1 KO mice were respectively pretreated with either vehicle or two doses (0.5 and 1.0 U/ml) of human thrombin (Sigma) for 24 h. Then, some cells were exposed to OGD for 3 h and astrocytic injury was quantified by lactate dehydrogenase (LDH) release assay and live/dead cell staining methods (n = 12 respectively) 24 h after OGD. Other cells were harvested immediately after pretreatment for Western blot analysis (n = 5) or immunocytochemistry to measure phosphorylated p44/42 MAPK and p90RSK as well as HSP25.
Part II. Expressions of phosphorylated p90RSK and HSP25 were examined in WT mouse astrocytes after TPC (0.5 U/ml, n = 8) with or without the pretreatment of PD98059 (an inhibitor of p44/42 MAPK; 20 μM, Calbiochem) for 30 minutes. Some cells were exposed to OGD for 3 h and astrocytic injury was quantified as described in Part I, while other cells were collected for Western blot analysis or immunocytochemistry to measure phosphorylated p90RSK (T359/S363) level and HSP25 expression.
Part III. Expression of HSP25 was examined in WT mouse astrocytes after TPC (0.5 U/ml, n = 8) with or without pretreatment with SL0101 (100 μM; an inhibitor of p90RSK, Calbiochem) for 3 h. Some cells were exposed to OGD for 3 h and astrocytic injury was quantified as described in Part I while other cells were collected for Western blot analysis or immunocytochemistry to measure HSP25 expression.
Oxygen glucose deprivation
Astrocytes were transferred into a temperature-controlled (37° ± 1°C) anaerobic chamber (Coy Laboratory, MI, U.S.A.) containing a gas mixture composed of 5% CO2, 10% H2, 85% N2. This chamber maintains a strict 0–5 ppm oxygen atmosphere through the hydrogen gas reacting with palladium catalyst to remove oxygen. The culture medium was replaced with deoxygenated glucose-free DMEM solution (Gibco), and astrocytes were maintained in the anaerobic chamber for 3 hours. After that, medium was replaced by serum-free medium and returned to a normoxic incubator under 5% CO2/ 95% air.
Measurement of lactate dehydrogenase
Astrocytic death was assessed by measurement of released LDH using a CytoTox-96 kit (Promega, Madison, WI, U.S.A.). Results were analyzed with microplate data analysis software, KC junior (BioTek Instruments, Inc.).
Live/dead cell determination
Cell viability was also measured by using a LIVE/DEAD Viability/Cytotoxicity assay kit (Molecular Probes, Eugene, OR, U.S.A.). Calcein-AM dyes living cells with bright-green fluorescence at excitation/emission wavelengths of 495/515 nm. The assay for dead cells used ethidium homodimer-1 (EthD-1), a molecule that binds to DNA and then fluoresces red at an excitation/emission wavelength of 495/635 nm. Briefly, the samples were washed 3 times in phosphate-buffered saline (PBS), and then were incubated with 2 μM calcein-AM and 4 μM EthD-1 in PBS for 30 minutes at room temperature. Quantification of dead cells (percent of red cells/red plus green cells) was performed by ImageJ 1.44 software.
Western blot analysis
Western blots were performed as described previously with some modification (Xi et al., 1999). Astrocytes were collected and immersed in Western sample buffer (62.5 mM Tris-HCl, pH 6.8; 2.3% sodium dodecyl sulfate; 10% glycerol; and 5% β-mercaptoethanol) and were then sonicated for 10 seconds. Protein concentrations were measured using a Thermo Scientific Pierce BCA Protein Assay Kit. Samples were loaded after 5 minutes boiling at 95°C. The protein was transferred to a Hybond-C pure nitrocellulose membrane (Amersham). The membranes were blocked in 5% Carnation non-fat dry milk for 2 hours at room temperature. Blots were washed and membranes were incubated with the following primary antibodies: polyclonal rabbit phospho-p44/42 MAPK antibody (1:2000; Cell Signaling, MA, U.S.A.), polyclonal rabbit p44/42 MAPK antibody (1:2000; Cell Signaling), polyclonal rabbit phospho-p90RSK (T359/S363) antibody (1:1000; Cell Signaling), polyclonal rabbit p90RSK antibody (1:1000; Cell Signaling) and polyclonal rabbit HSP27 antibody (1:1000; Cell Signaling). The appropriate second antibodies were applied for 1 hour at room temperature. Proteins were visualized with the ECL chemiluminescence system (Amersham) and exposed to Kodak X-Omat film. The relative densities of the protein bands were analyzed with ImageJ 1.44 software.
Immunocytochemistry
Astrocytes were grown on chamber slides and fixed with 4% paraformaldehyde. Slides were rinsed and the cells permeabilized with 100% methanol at -20°C for 5 minutes. Polyclonal rabbit phospho-p44/42 MAPK antibody (1:400), polyclonal rabbit phospho-p90RSK (T359/S363) antibody (1:200) and polyclonal rabbit HSP27 antibody (1:200; Cell Signaling) were applied and the slides incubated overnight at 4°C in a moist chamber after 10% normal goat serum blocking for 1 hour at room temperature. After washing three times, the slides were incubated with rhodamine goat anti-rabbit IgG (1:500, Chemico, Temecula, CA, U.S.A.) for 1 hour at room temperature, and astrocytes were then photographed using a fluorescence microscope.
Statistical analysis
All data in this study are presented as mean ± standard deviation. Data were analyzed with Student’s t test or ANOVA test. Significance levels were set at p < 0.05.
Results
Effects of TPC on astrocytic death induced by OGD in WT and PAR-1 KO groups
Cultured mouse astrocytes were pretreated with two low doses of thrombin (0.5 and 1.0 U/ml) for 24 hours. Cells then underwent 3 hours OGD, and both LDH assays were performed 24 hours after OGD. LDH assay showed that WT mouse astrocytes pretreated with 0.5 and 1.0 U/ml thrombin had significantly lower LDH release compared to no TPC [49.9 ± 13.1% (thrombin 0.5 U/ml), 59.6 ± 9.2% (thrombin 1.0 U/ml) vs. control (100%), p < 0.01]. In contrast, there was no difference between TPC groups and the non-preconditioning group in PAR-1 KO astrocytes [94.7 ± 17.9% (thrombin 0.5 U/ml), 92.9 ± 19.7% (thrombin 1.0 U/ml) vs. control (100%)] (Fig. 1A).
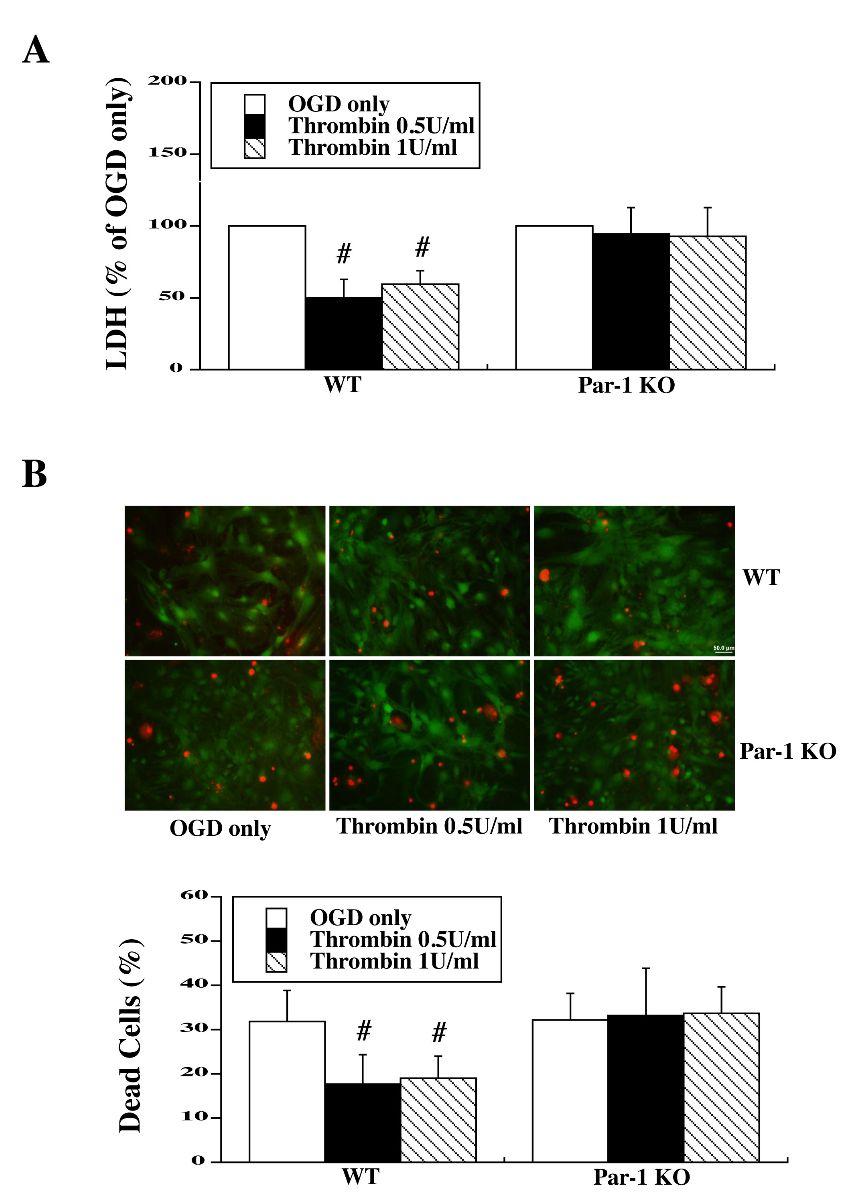
In a new window | Download PPT
Figure 1: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with two doses of thrombin for 24 h and then exposed to oxygen glucose deprivation (OGD) for 3 h. (A) The levels of lactate dehydrogenase (LDH) released into the medium and (B) the percent of dead cells by live/dead cell staining were measured after 24 h of reoxygenation. Values are expressed as means ± SD, #p < 0.01 vs. OGD only (0 U/ml thrombin). In (B), astrocytes stained red indicate dead and those green are live. Scale bar = 50 μm.
Similar results were obtained with live/dead cell staining 24 hours after OGD. TPC reduced cell death in WT astrocytes [17.6 ± 6.8% dead (WT; thrombin 0.5 U/ml), 19.0 ± 5.1% (WT; thrombin 1.0 U/ml) vs. 31.8 ± 7.1% in control; p < 0.01). In contrast, there was no significant difference in cell death with TPC in PAR-1 KO astrocytes [33.3 ± 10.6% dead (KO thrombin 0.5 U/ml), 33.6 ± 6.1% (KO thrombin 1.0 U/ml) vs. 32.3 ± 5.9% in control] (Fig. 1B).
Role of PAR-1 in TPC-induced p44/42 MAPK/ p90RSK pathway activation and HSP25 upregulation
Astrocytes from WT and PAR-1 KO mice were used to examine the role of PAR-1 in the activation of the p44/42 MAPK/p90RSK (T359/S363) pathway by TPC. Both immunohistochemistry (Fig. 2A; 3A) and Western blot (Fig. 2B; 3B) showed that TPC (0.5 or 1 U/ml) phosphorylated p44/42 MAPK and the downstream p90RSK (T359/S363) in WT astrocytes. No significant upregulation of phosphorylated p44/42 MAPK and p90RSK was found in PAR-1 KO astrocytes (Fig. 2; 3).

In a new window | Download PPT
Figure 2: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with two doses of thrombin (0.5 and 0.1 U/ml) for 24 h and then collected for immunofluorescence staining and Western blot analysis. (A) Phosphorylated p44/42 MAPK immunoreactivity in astrocytes 24 h after control or thrombin (0.5, 1.0 U/ml) treatment. (B) Western blot showing phosphorylated (P-) and total (T-) p44/42 MAPK protein levels in astrocytes after control or thrombin treatment for 24 h. Values are mean ± SD, *p < 0.05 vs. the WT control group and KO TPC group. Scale bar = 50 μm.
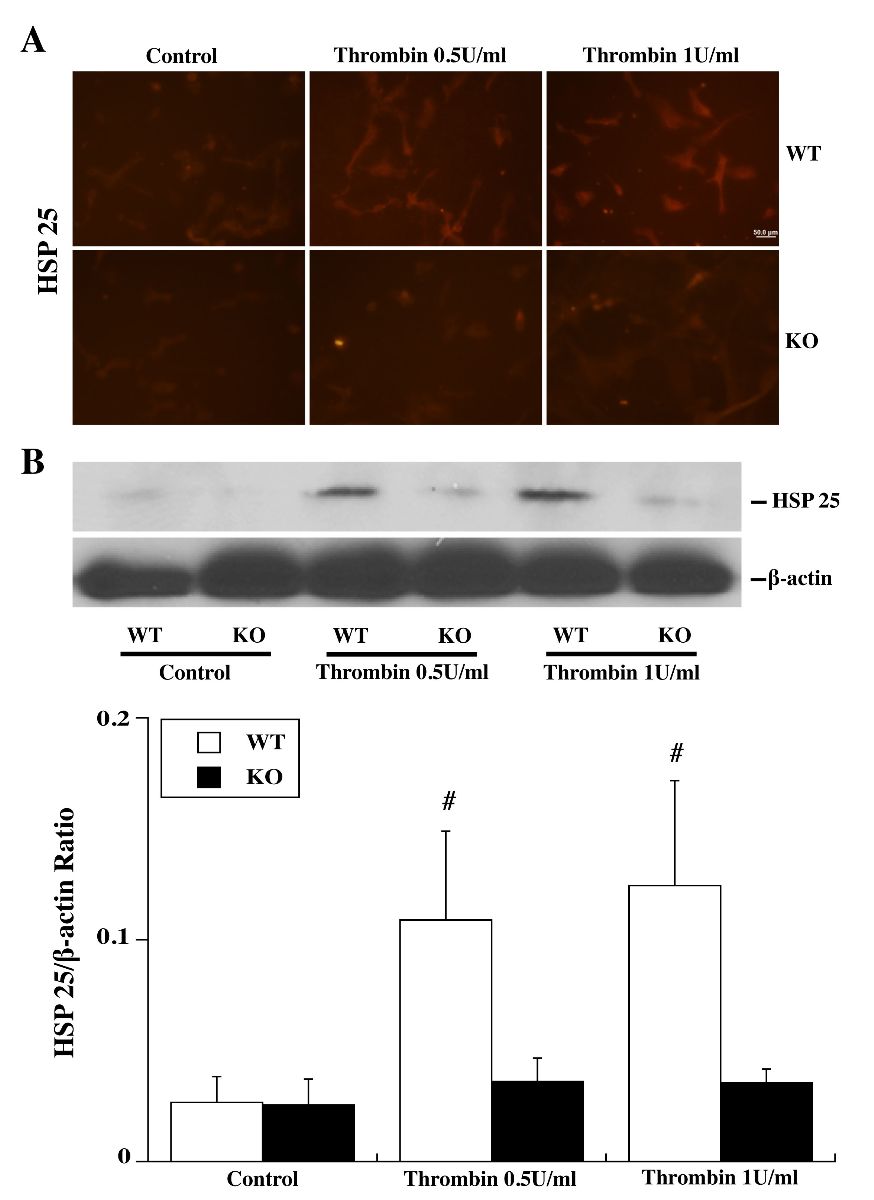
TPC induced a marked upregulation in HSP25 in WT astrocytes as assessed by immunohistochemistry (Fig. 4A) and Western Blot (Fig. 4B). Again, this upregulation was absent in PAR-1 KO astrocytes (Fig. 4).
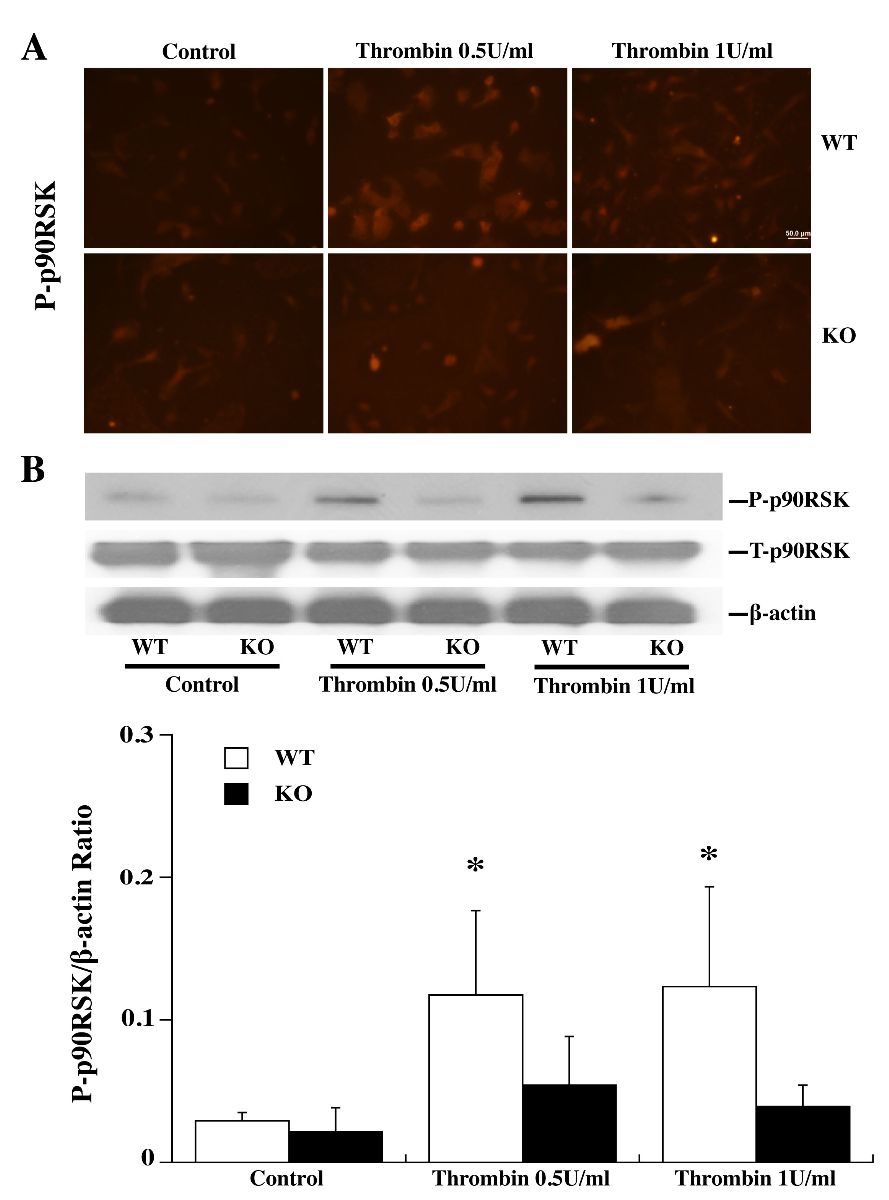
In a new window | Download PPT
Figure 3: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with 0 (control), 0.5 or 1.0 U/ml thrombin for 24 h and then collected for immunofluorescence staining and Western blot analysis. (A) Immunoreactivity of phosphorylated p90RSK (T359/S363) in astrocytes 24 h after control or thrombin treatment. (B) Western blots showing phosphorylated (P-) and total (T-) p90RSK protein levels in astrocytes after control or thrombin treatment. Values are mean ± SD, *p < 0.05 vs. the control and KO thrombin groups. Scale bar = 50 μm.
Effects of a p44/42 MAPK pathway inhibitor, PD98059, on TPC-induced protection and upregulation of phosphorylated p90RSK and HSP25
To examine the hypothesis that the protective effects of TPC in WT astrocytes are mediated by activating the p44/42 MAPK pathway, an inhibitor of that pathway, PD98059, was added 30 minutes before TPC (0.5 U/ml). Twenty-four hours later, the astrocytes were exposed to OGD for 3 hours, and LDH levels and live/dead cells were examined after a further 24 hours. PD98059 abolished TPC-induced protection against OGD (Fig. 5A, 5B).
In a new window | Download PPT
Figure 4: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with 0 (control), 0.5 or 1.0 U/ml thrombin for 24 h and then collected for immunofluorescence staining and Western blot analysis. (A) HSP25 immunoreactivity in astrocytes after control or thrombin treatment. (B) Western blot showing HSP25 protein levels in astrocytes after control or thrombin treatment. Values are mean ± SD, #p < 0.01 vs. control group and KO thrombin treatment group. Scale bar = 50 μm.
Astrocytes were also collected immediately at 24 hours after TPC for immunofluorescent and Western blot analysis. PD98059 blocked the phosphorylation of p90RSK (T359/S363) induced by TPC (Fig. 5C). Similarly, it blocked the upregulation of HSP25 found with TPC (Fig. 5C).
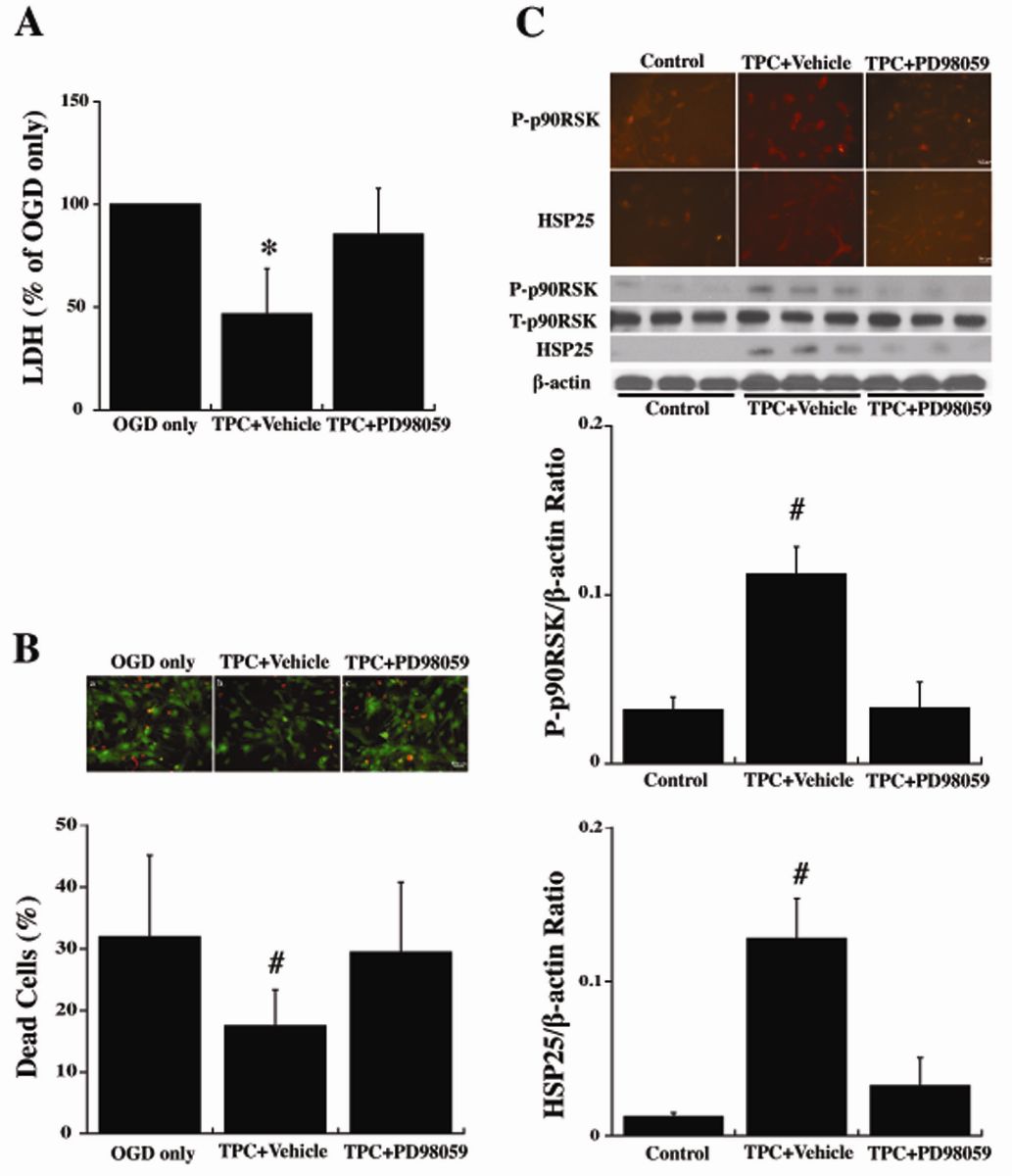
In a new window | Download PPT
Figure 5: Primary cultured WT mouse astrocytes were pretreated with PD98059 (20 μM) an inhibitor of p44/42 MAPK, or vehicle for 30 min before 24 hours thrombin (0.5 U/ml) treatment (TPC). Astrocytes were then exposed to OGD for 3 h and (A) LDH levels and (B) the % of dead cells was measured after 24 h of reoxygenation. (C) Other astrocytes were collected after the 24 hours of thrombin treatment to assess immunoreactivity and protein levels (Western blot) of total (T-) and phosphorylated (P-)p90RSK (T359/S363) and HSP25. Values are mean ± SD, for control (0 U/ml thrombin), TPC + vehicle and TPC + PD98059 groups. *p < 0.05, #p < 0.01 indicate TPC + vehicle vs. the other groups. Scale bar = 50 μm.
Effects of a p90RSK inhibitor, SL0101, on TPC-induced protection and HSP25 upregulation
To examine the hypothesis that the protective effects of TPC and p44/42 MAPK pathway activation are mediated by activating p90RSK, an inhibitor, SL0101, was added 30 minutes before TPC (0.5 U/ml). Twenty-four hours later, the astrocytes were exposed to OGD for 3 hours, and LDH levels and live/dead cells were examined after a further 24 hours. SL0101 blocked TPC-induced protection against OGD (Fig. 6A, 6B).
Astrocytes were also collected immediately at 24 hours after TPC for immunofluorescent and Western blot analysis. SL0101 reduced the upregulation of HSP25 found with TPC (Fig. 6C).
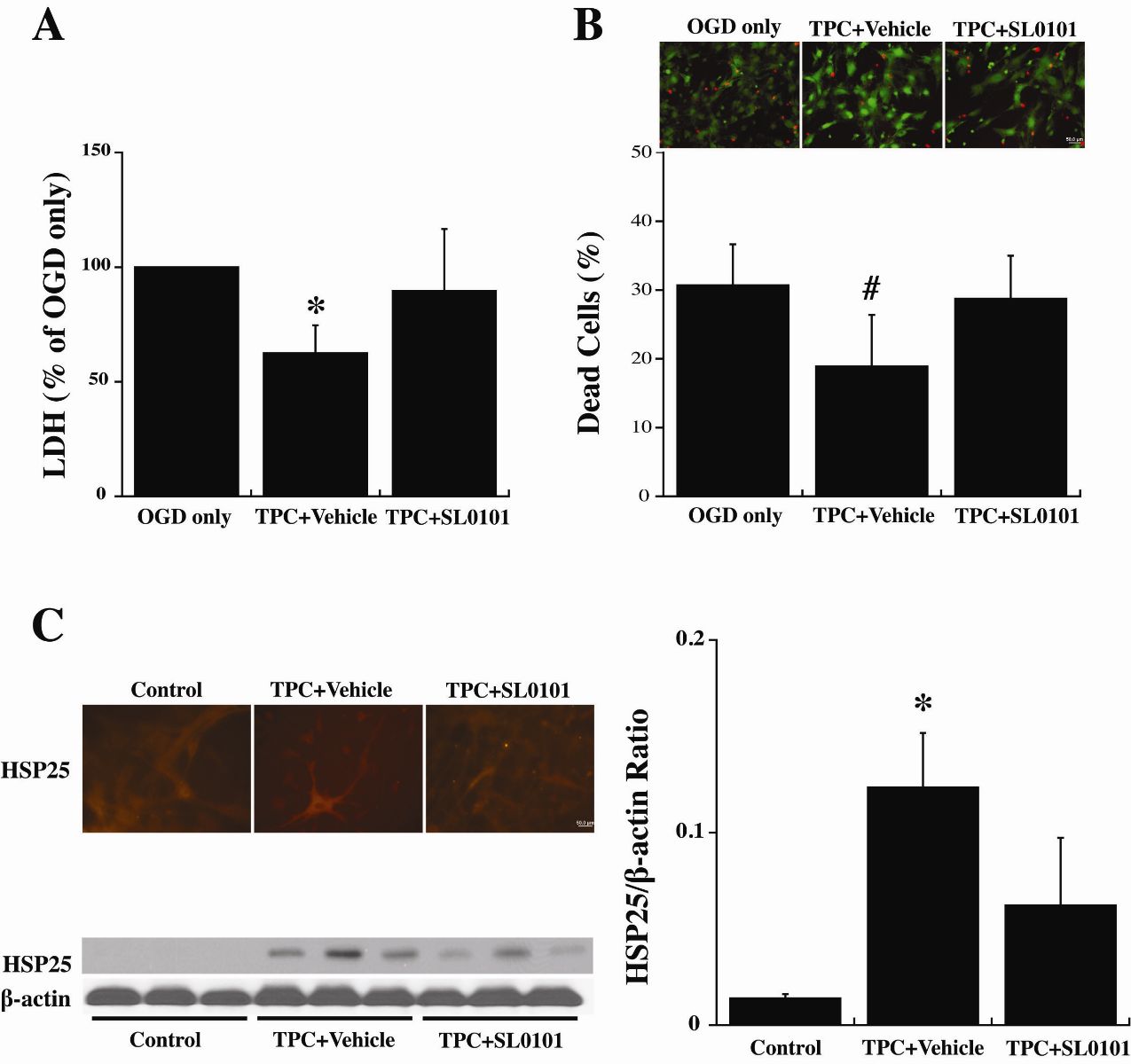
In a new window | Download PPT
Figure 6: Primary cultured WT mouse astrocytes were pretreated with SL0101 (100 μM), an inhibitor of p90RSK, or vehicle for 3 hours before 24 hours thrombin (0.5 U/ml) treatment (TPC). Astrocytes were then exposed to OGD for 3 h and (A) LDH levels and (B) the % of dead cells was measured after 24 h of reoxygenation. (C) Other astrocytes were collected after the 24 hours of thrombin treatment to assess immunoreactivity and protein levels of HSP25. Values are mean ± SD, for control (0 U/ml thrombin), TPC + vehicle and TPC + SL0101 groups. *p < 0.05, #p < 0.01 indicate TPC + vehicle vs. the other groups. Scale bar = 50 μm.
Discussion
This study reports several important findings: 1) Thrombin preconditioning exerts protection against astrocytic death induced by OGD only in WT mouse astrocytes and has nearly no protective effect in PAR-1 KO mouse astrocytes. 2) Thrombin preconditioning upregulated phosphorylated p44/42 MAPK, phosphorylated p90RSK (T359/S363) and HSP25 levels in WT mouse astrocytes but not in PAR-1 KO astrocytes. 3) PD98059, an inhibitor of p44/42 MAPK, not only reduced TPC-induced protection, it also blocked the upregulation of phosphorylated p90RSK (T359/S363) and HSP25 levels. 4) SL0101, an inhibitor of p90RSK, not only reduced protection induced by thrombin preconditioning, it also blocked upregulation of HSP25 levels. These results suggest that PAR-1 and the p44/42 MAPK/p90RSK/HSP25 pathway have an important role in thrombin-induced protection. It should be noted, however, that one limitation of this study is the lack of using the kinase inhibitors alone followed by OGD.
A variety of conditioning stimuli induce multiple endogenous protective mechanisms to limit different kinds of brain injury in preclinical models (Hu et al., 2010; Dirnagl et al., 2003; Gidday, 2016; Hirt et al., 2009; Keep et al., 2014; Guan et al., 2016). Conditioning stimuli have been examined in multiple clinical trials to limit brain and heart injury, although the results are still inconclusive (Hausenloy et al., 2015; Benstoem et al., 2017; Wang et al., 2017; Zhao et al., 2017). Chan et al. have reported application of ischemic preconditioning during clipping of cerebral aneurysm and found it attenuated tissue hypoxia (Chan et al., 2005). It is well known that neuroprotection and how to achieve it are always major events in neurosurgery since neurosurgical procedures may lead to brain edema, hemorrhage and/or ischemia. We have demonstrated that TPC can reduce hemorrhagic and ischemic brain injury both in vivo and in vitro (Xi et al., 2003). Therefore, activation of the pathways involved in TPC may be a method of protecting the brain during neurosurgical procedures.
The current study indicates that thrombin-induced ischemic tolerance in mouse astrocytes is initiated through PAR-1 and its downstream mechanisms. Protease-activated receptor-1 was the first discovered member of a unique class of G-protein-coupled signaling receptors that become activated via cleavage of their extracellular amino terminus by proteinases such as thrombin (Flynn and Buret, 2004). Striggow et al. have shown that the protective effects of thrombin are caused only by concentrations in the picomolar range and are mediated by PAR-1 activation (Striggow et al., 2000). They also found that PAR-1 was upregulated in hippocampus by severe ischemia in organotypic hippocampal slice cultures (Striggow et al., 2001). In our previous study, we found PAR-1 mRNA expression was upregulated after thrombin pretreatment in rat neurons and that a PAR-1 agonist markedly reduced OGD-induced neuronal death (Hu et al., 2010). In the present study, we found that TPC only exerted protective effects against OGD in astrocytes with PAR-1. The downstream MAPK activation and upregulation of HSP25 was also only observed in WT and not in PAR-1 KO astrocytes. Hence, PAR-1 plays an important role in initiating the cytoprotective effect induced by thrombin in multiple cell types.
The current study examined whether thrombin-induced protection is associated with the activation of p44/42 MAPK and the downstream mechanisms. MAPKs are well-known cytoplasmic signal transducers with an important role in both thrombin- and ischemia-induced brain tolerance (Xi et al., 2001; Irving and Bamford, 2002). As a major MAPK member, the p44/42 MAPK signaling pathway regulates a number of cellular process including cell differentiation, growth, survival and apoptosis (Pearson et al., 2001). In vivo, p44/42 MAPK are activated in the brain after intracerebral injection of thrombin, and PD98059 abolishes thrombin-induced activation of p44/42 MAPK and also blocks thrombin-induced brain tolerance (Xi et al., 2001). Moreover, thrombin increases brain hypoxia-inducible factor-1 alpha levels through a p44/42 MAPK pathway (Hua et al., 2003). In vitro, thrombin treatment activates p44/42 MAPK in both astrocytes and neurons (Hu et al., 2010; Wang et al., 2002), and PD98059 blocks the cytoprotective effect of thrombin pretreatment in mixed glial/neuronal cultures or pure neuronal cultures (Hu et al., 2010; Jiang et al., 2002). In combination with the current studies, these findings imply that TPC has effects on multiple cell types from different strains (at least rat neurons and astrocytes, as well as mouse astrocytes), through a common intracellular pathway, p44/42 MAPK signaling, to induce protection.
As a member of the kinase cascade, activation of p44/42 MAPK leads to the further activation of downstream kinases. Among the substrates of p44/42 MAPK are the family of p90 kDa ribosomal S6 kinases (p90RSK) (Frodin and Gammeltoft, 1999). p90RSK is involved in a number of cell functions including regulating gene expression and cell cycle, phosphorylation of downstream proteins, and induction of new protein synthesis. The latter may be directed by activation of a variety of transcription factors, such as polyribosomal proteins and glycogen synthase kinase-3 (Frodin and Gammeltoft, 1999; Anjum and Blenis, 2008). A previous study showed p90RSK was involved in the protective effect of melatonin against ischemic brain injury (Koh, 2008). There are several phosphorylation sites on p90RSK, including Ser380, Thr573 and Thr359/Ser363 that regulate function (Anjum and Blenis, 2008). For example, the Ser380 site serves as a hydrophobic motif, while phosphorylation of the Thr573 site may contribute to kinase translocation to the plasma membrane (Anjum and Blenis, 2008). However, as yet, the functional effects of p90RSK phosphorylation, including at Thr359/Ser363, are not well known (Anjum and Blenis, 2008). The current study found that TPC upregulated the phosphorylation of p90RSK (T359/S363) via activation of the PAR-1/p44/42 MAPK pathway. Inhibiting p90RSK with SL0101 blocked both TPC-induced HSP25 upregulation and the protection against astrocytic death after OGD. These results imply that phosphorylation of Thr359 and Ser 363 sites upon p90RSK may be associated with the protein synthesis leading to cell survival.
New protein synthesis is crucial for preconditioning-induced neuroprotection (Dirnagl et al., 2009). Our previous studies also found that thrombin-induced protection may involve the induction of heat shock proteins, particularly HSP27 (called HSP25 in mouse) in rat models (Xi et al., 1999). HSP25/27 is reported to perform many cellular protective functions. These include mediating cell survival directly or undergoing recognition and chaperoning of damaged or misfolded proteins (Stetler et al., 2009). HSP27 also protects against heat shock and oxidative stress by stabilizing actin filaments, which is related to activation of a stress-sensitive MAPK signal transduction pathway that induces resistance to stress-induced actin fragmentation (Xi et al., 1999). HSP27 protects against ischemic brain injury via attenuation of a novel stress-response cascade upstream of mitochondrial cell death signaling and attenuation of blood-brain barrier disruption (Stetler et al., 2008; Shi et al., 2017). Overexpression of HSP27 (transgenic mice) reduces cortical damage after cerebral ischemia (van der Weerd et al., 2010). These results, along with our finding that PD98059 and SL0101 both prevented TPC-induced HSP25 upregulation and protection against OGD, suggest that HSP25 may have a crucial role in TPC-induced protection. This merits further investigation.
Apart from relevance to therapeutic preconditioning, the current results also raise the question of whether PAR-1 activation following microhemorrhages (and associated thrombin production) might have protective effects. It will be important to determine whether such hemorrhages activate the p44/42 MAPK/p90RSK/HSP27 pathway and how long that activation persists.
In conclusion, our results show that thrombin preconditioning can have direct effects on astrocytes to induce protection against ischemia-like conditions. Moreover, they also indicate that activation of PAR-1 and the p44/42 MAPK/p90RSK/HSP25 pathway may play a key role in that astrocytic protection.
Acknowledgements
This study was supported by grants NS-073595, NS-091545, NS-090925, and NS-096917 from the National Institutes of Health (NIH).
References
Xuhui Bao
1Department of Neurosurgery, University of Michigan, Ann Arbor, MI, USA
Ya Hua
1Department of Neurosurgery, University of Michigan, Ann Arbor, MI, USA
Richard F. Keep
1Department of Neurosurgery, University of Michigan, Ann Arbor, MI, USA
Guohua Xi
1Department of Neurosurgery, University of Michigan, Ann Arbor, MI, USA
Corresponding author:
Guohua Xi
Email: guohuaxi@umich.edu
In a new window | Download PPT
Figure 1: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with two doses of thrombin for 24 h and then exposed to oxygen glucose deprivation (OGD) for 3 h. (A) The levels of lactate dehydrogenase (LDH) released into the medium and (B) the percent of dead cells by live/dead cell staining were measured after 24 h of reoxygenation. Values are expressed as means ± SD, #p < 0.01 vs. OGD only (0 U/ml thrombin). In (B), astrocytes stained red indicate dead and those green are live. Scale bar = 50 μm.
In a new window | Download PPT
Figure 2: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with two doses of thrombin (0.5 and 0.1 U/ml) for 24 h and then collected for immunofluorescence staining and Western blot analysis. (A) Phosphorylated p44/42 MAPK immunoreactivity in astrocytes 24 h after control or thrombin (0.5, 1.0 U/ml) treatment. (B) Western blot showing phosphorylated (P-) and total (T-) p44/42 MAPK protein levels in astrocytes after control or thrombin treatment for 24 h. Values are mean ± SD, *p < 0.05 vs. the WT control group and KO TPC group. Scale bar = 50 μm.
In a new window | Download PPT
Figure 3: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with 0 (control), 0.5 or 1.0 U/ml thrombin for 24 h and then collected for immunofluorescence staining and Western blot analysis. (A) Immunoreactivity of phosphorylated p90RSK (T359/S363) in astrocytes 24 h after control or thrombin treatment. (B) Western blots showing phosphorylated (P-) and total (T-) p90RSK protein levels in astrocytes after control or thrombin treatment. Values are mean ± SD, *p < 0.05 vs. the control and KO thrombin groups. Scale bar = 50 μm.
In a new window | Download PPT
Figure 4: Primary astrocyte cultures from WT and PAR-1 KO mice were pretreated with 0 (control), 0.5 or 1.0 U/ml thrombin for 24 h and then collected for immunofluorescence staining and Western blot analysis. (A) HSP25 immunoreactivity in astrocytes after control or thrombin treatment. (B) Western blot showing HSP25 protein levels in astrocytes after control or thrombin treatment. Values are mean ± SD, #p < 0.01 vs. control group and KO thrombin treatment group. Scale bar = 50 μm.
In a new window | Download PPT
Figure 5: Primary cultured WT mouse astrocytes were pretreated with PD98059 (20 μM) an inhibitor of p44/42 MAPK, or vehicle for 30 min before 24 hours thrombin (0.5 U/ml) treatment (TPC). Astrocytes were then exposed to OGD for 3 h and (A) LDH levels and (B) the % of dead cells was measured after 24 h of reoxygenation. (C) Other astrocytes were collected after the 24 hours of thrombin treatment to assess immunoreactivity and protein levels (Western blot) of total (T-) and phosphorylated (P-)p90RSK (T359/S363) and HSP25. Values are mean ± SD, for control (0 U/ml thrombin), TPC + vehicle and TPC + PD98059 groups. *p < 0.05, #p < 0.01 indicate TPC + vehicle vs. the other groups. Scale bar = 50 μm.
In a new window | Download PPT
Figure 6: Primary cultured WT mouse astrocytes were pretreated with SL0101 (100 μM), an inhibitor of p90RSK, or vehicle for 3 hours before 24 hours thrombin (0.5 U/ml) treatment (TPC). Astrocytes were then exposed to OGD for 3 h and (A) LDH levels and (B) the % of dead cells was measured after 24 h of reoxygenation. (C) Other astrocytes were collected after the 24 hours of thrombin treatment to assess immunoreactivity and protein levels of HSP25. Values are mean ± SD, for control (0 U/ml thrombin), TPC + vehicle and TPC + SL0101 groups. *p < 0.05, #p < 0.01 indicate TPC + vehicle vs. the other groups. Scale bar = 50 μm.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11220 | 13 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA