Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Differential regulation of cerebral microvascular transcription by single and repetitive hypoxic conditioning
Time:2021-04-05
Number:8109
Author Affiliations

Conditioning Medicine 2021. 4(1):58-68.
Abstract
Systemic conditioning therapeutics afford brain protection at all levels of organization, occurring autonomously for neurons, glia, vascular smooth muscle, and endothelium, which are mediated systemically for the adaptive and innate immune system. The present study was undertaken to examine acute (3 h) and delayed (2 days) gene expression changes in mouse cerebral microvessels following single hypoxic conditioning (HX1) and repetitive hypoxic conditioning (HX9), the latter for which we showed previously to extend focal stroke tolerance from days to months. Microarray (Illumina) analyses were performed on microvessel-enriched fractions of adult mouse brain obtained from the following five groups (naïve; HX1-3h; HX1-2days; HX9-3h; HX9-2days). Differentially expressed genes were analyzed bioinformatically using Ingenuity Pathway Analysis software, with qPCR validating selected up- and down-regulated genes. As expected, some differentially expressed genes were common to more than one treatment or time point, whereas others were unique to treatment or time point. Bioinformatic analyses provided insights into acute (3h) inflammatory and immune signaling pathways that may be differentially activated by HX1 and HX9, with anti-inflammatory and trophic pathways coincident with the ischemia-tolerant phenotype two days after HX1. Interestingly, two days after HX9, microvessels were transcriptionally silent, with only five genes remaining differentially expressed relative to naïve mice. Our microarray findings and bioinformatic analyses suggest that cerebral microvessels from HX1-treated mice exhibit early activation of immune system signaling that is largely suppressed in microvessels from HX9-treated mice. These and other differences between these responses require further study, including at the proteomic level, and with pharmacologic and genetic experiments designed to reveal causality, to reveal further insights into the mechanisms underlying long-lasting stroke tolerance.
Keywords: Preconditioning, hypoxia, microcirculation, microarray, vascular protection,neuroprotection, epigenetics, bioinformatics, resilience
Abstract
Systemic conditioning therapeutics afford brain protection at all levels of organization, occurring autonomously for neurons, glia, vascular smooth muscle, and endothelium, which are mediated systemically for the adaptive and innate immune system. The present study was undertaken to examine acute (3 h) and delayed (2 days) gene expression changes in mouse cerebral microvessels following single hypoxic conditioning (HX1) and repetitive hypoxic conditioning (HX9), the latter for which we showed previously to extend focal stroke tolerance from days to months. Microarray (Illumina) analyses were performed on microvessel-enriched fractions of adult mouse brain obtained from the following five groups (naïve; HX1-3h; HX1-2days; HX9-3h; HX9-2days). Differentially expressed genes were analyzed bioinformatically using Ingenuity Pathway Analysis software, with qPCR validating selected up- and down-regulated genes. As expected, some differentially expressed genes were common to more than one treatment or time point, whereas others were unique to treatment or time point. Bioinformatic analyses provided insights into acute (3h) inflammatory and immune signaling pathways that may be differentially activated by HX1 and HX9, with anti-inflammatory and trophic pathways coincident with the ischemia-tolerant phenotype two days after HX1. Interestingly, two days after HX9, microvessels were transcriptionally silent, with only five genes remaining differentially expressed relative to naïve mice. Our microarray findings and bioinformatic analyses suggest that cerebral microvessels from HX1-treated mice exhibit early activation of immune system signaling that is largely suppressed in microvessels from HX9-treated mice. These and other differences between these responses require further study, including at the proteomic level, and with pharmacologic and genetic experiments designed to reveal causality, to reveal further insights into the mechanisms underlying long-lasting stroke tolerance.
Keywords: Preconditioning, hypoxia, microcirculation, microarray, vascular protection,neuroprotection, epigenetics, bioinformatics, resilience
Introduction
Preconditioning elicits robust protection of the brain from ischemic and other injuries by epigenetically mediated changes in gene expression (Thompson et al., 2013; Gidday, 2015; Cavalli and Heard, 2019). However, the transient nature of these adaptive responses limit direct clinical translation; even post-conditioning requires rapid post-injury application. Studies from our lab and others over the last decade confirmed that repetitive conditioning can extend the duration of ischemic tolerance so induced from days to weeks, sometimes months, after the cessation of treatment (Stowe et al., 2011; Lovett-Barr et al., 2012; Gidday, 2015). The mechanisms by which sustained changes in phenotype result from repeated conditioning remain largely undefined (Leak et al., 2018). Given that systemic conditioning therapies induce adaptive epigenetic changes in all cells of the brain – with considerable evidence for direct (Stowe and Gidday, 2013) and indirect (Xing and Lo, 2017) cerebrovascular protection – we undertook the present study to investigate the transcriptional responses of the cerebral microcirculation to single and repetitive conditioning, and then subjected these results to extensive bioinformatic analyses, to gain insights into the vascular mechanisms potentially underlying the distinct durations of ischemic tolerance they respectively induce.
Materials and Methods
All procedures were approved by Washington University’s Institutional Animal Care and Use Committee, and adhered to ARRIVE guidelines, and the NIH Guide for the Care and Use of Laboratory Animals.
Hypoxic preconditioning treatments and experimental groups. Previous studies from our lab consistently confirmed that a single systemic hypoxia challenge (HX1) induced transient periods of neurovascular protection in the brain (Miller et al., 2001; Stowe et al., 2012) for 1-3 days, whereas repetitive hypoxic conditioning protocols extended the duration of ischemic tolerance for up to two months thereafter (Stowe et al., 2011; Selvaraj et al., 2017). Thus, we randomly divided animals into four age-matched experimental cohorts, plus a naïve normoxic control cohort not subjected to hypoxic conditioning.
Outbred male Swiss-Webster ND4 mice, 8-12 wks old, were obtained from Envigo (Indianapolis, IN, USA) and maintained on a 12-h light/dark cycle with chow provided ad libitum. Following a 2-wk period of acclimation to our institutional vivarium, our hypoxic preconditioning treatments were initiated. HX1 involved a single exposure of mice to systemic hypoxia (11% oxygen) for 4 h. HX9 involved exposing mice to two levels of systemic hypoxia (11% or 8%) for 2 or 4 h, as described previously (Stowe et al., 2011). Subgroups of HX1- and HX9-treated mice were then randomized to be sacrificed, alongside age-matched, untreated naïve controls, either 3 h or 2 days following the single hypoxic exposure in the HX1 group, or 3 h or 2 days following the final hypoxic exposure in the HX9 group, for microarray analyses by a blinded observer.
Microvessel isolation. To elucidate temporal changes in endothelial cell gene expression in response to our single and repetitive hypoxic stimuli, microvessels (including capillaries, small arterioles, and venules) were isolated from flash-frozen neocortex via differential centrifugation in sucrose buffer from ipsilesional hemispheres, as performed previously by our lab (Wacker et al. 2009; Stowe et al., 2011) based on the protocol originally detailed by the Lester Drewes laboratory (Gerhart et al. 1988). In brief, cortical hemispheres were flash-frozen after transcardiac perfusion with 0.1 M phosphate buffered saline (PBS), and all Eppendorfs and pipette tips were coated with BSA prior to each step. Each hemisphere was hand-homogenized in 1 ml sucrose/PI buffer (0.32 M sucrose, 3 mM HEPES, RNAse-free water (pH 7.4) with 0.05% phosphatase inhibitor (Activ Motif, Carlsbad, CA) prior to centrifugation (10 min, 4 °C, 1000g). The pellet was resuspended in 850 µl sucrose/PI buffer, centrifuged (15 sec., 4 °C, 100g), and 720 µl of the supernatant was removed, and placed in a 2-ml BSA-coated tube on ice. The pellet was resuspended in 850 µl sucrose/PI buffer, centrifuged again (15 sec., 4 °C, 100g), and the resultant supernatant was added to the previously collected supernatant, centrifuged briefly (1 min., 4 °C, 200g), and 1ml of supernatant was removed and resuspended in 1.25 ml sucrose/PI buffer, and centrifuged (2 min., 4 °C, 200g) to obtain the microvessel-rich pellet. The pellet was resuspended in 0.01 M RNAse-free PBS, centrifuged again (3 min., 4 °C, 9000g), and all PBS discarded. Pellets were stored at -80 °C until processing.
RNA isolation and qPCR. Total RNA preparations were obtained using an established Trizol protocol, as described previously (Selvaraj et al., 2017). After DNase I treatment (Invitrogen, Carlsbad, CA), total RNA was then quantified using the NanoDrop method (Thermo Scientific, Wilmington, DE). Quantitative real-time PCR was performed as described (Selvaraj et al., 2017) to validate, at the mRNA level, selected genes that exhibited differential expression (both up- and down-regulation) in response to HX1 or HX9. Student’s t-test determined significance.
Microarray analysis. Total RNA was analyzed on Illumina Mouse-6 chips. Detectable genes in microvessels from each of the four experimental groups were normalized to the same genes in microvessel preps obtained from naïve mice using Partek Genomics Suite, with final between-group significance of the 18,099 genes detected determined via restricted maximum likelihood (REML) ANOVA. Bioinformatic analysis of microarray data was performed following acquisition as described below.
Bioinformatic analyses. Array-acquired data were analyzed using Qiagen’s Ingenuity Pathway Analysis (IPA) software. Except for the HX9-2 day group, in which only five differentially expressed genes were found, a ‘Core Analysis’ was performed using only the genes found in each of the three remaining experimental groups that were differentially expressed relative to the non-conditioned naïve group to identify the top Canonical Pathways, Diseases & Disorders, and Cellular & Molecular Functions that define each respective group, and to identify the top Upstream Regulators of these various pathways and networks predicted for each group. We subsequently performed a ‘Comparison Analysis’ between the three experimental groups to reveal secondary lists of top Canonicals, Biological Functions, and Upstream Regulators for each group based on the differentially expressed genes that are shared among them, and obtained the associated heat maps for every Canonical, Function, and Regulator in each group that reveal, by z-score, the overall level of activation or inhibition of that particular pathway/network and regulator in that group.
In all of our bioinformatics analyses, a minimum z-score threshold of > ± 2 was applied, which represents a p < 0.05 equivalent metric for the non-randomness of directionality of a given dataset. IPA Analysis content information: Report Date: 12/24/2020; Content Version: 60467501 (Release Date: 11/19/2020); HX 1 3h Report ID: 24439872; HX 1 2d Report ID: 24439912; HX 9 3h Report ID: 24439932. Array data were displayed using GraphPad 9 data visualization software (v9.0.0 San Diego, CA). Figures 4 and 5 were created using BioRender (www.biorender.com).
Results and Discussion
Systemic conditioning stimuli, including hypoxia, affect all resident brain cells, as well as circulating immune cells. With respect to the cerebral circulation, not only do endothelial cells exhibit adaptive epigenetic responses to preconditioning for autonomous protection from injury, but they also serve as cellular intermediaries for systemic immune responses. We previously showed that the cerebrovasculature reacts to conditioning stimuli-induced environmental (i.e., systemic) changes by directly altering selectin and integrin expression on its luminal surface, by releasing paracrine mediators abluminally that induce adaptive epigenetic responses on the part of surrounding neurons and glia, and by altering chemokine expression required for immune cell diapedesis into the parenchyma (Stowe et al., 2012; Selvaraj et al., 2017). These and other pathways initiated by many systemic conditioning paradigms render interpretations of genome-wide responses of the cerebral microcirculation to systemic conditioning therapeutics challenging. The transcriptional changes we noted in the present study are no exception. That said, some clarity is provided by the time point at which the genomic response is probed, and the power of bioinformatic analyses to delineate transcriptional responses reflecting signaling from those gene expression changes associated with maintaining a steady-state, ischemia-tolerant phenotype. Below, we share our microarray results and predicted outcomes of our bioinformatic analyses of two distinct conditioning treatments, at two distinct time points, and their potential implications with respect to beginning to understand each respective cerebral microvascular conditioning response.
Overall Cerebral Microvascular Genomic Response and qPCR Validation. In total 18,099 genes were detected, of which 881 were significantly regulated in the microvessels of the adult mouse brain after either HX1 or HX9. Figure 1 shows the microarray data for the differentially expressed genes shared between either hypoxia treatments or time points. With respect to genes uniquely induced by treatment, a total of 236 genes were uniquely expressed 3 h after the HX1 conditioning stimulus: 119 upregulated and 117 downregulated (Figure 1A). Two days after HX1 conditioning, 235 genes were uniquely expressed at this time point (162 upregulated and 128 downregulated). A similar robust response in gene regulation was found 3 h after HX9, with a total of 235 genes uniquely expressed, 119 of which were upregulated and 116 downregulated (Figure 1C). However, 2 days after the HX9 conditioning treatment, differential gene regulation was greatly suppressed, with only 2 unique genes identified: 1 upregulated, pre-mRNA processing factor 8 (Prpf8) (1.3-fold) and 1 downregulated, D930020N02Rik (-1.1-fold). The majority of the differentially expressed genes in our study were unique to the specific sampling time points of either 3h or 2d, with only 18 genes common to both post-treatment time points (11 up, 7 down).
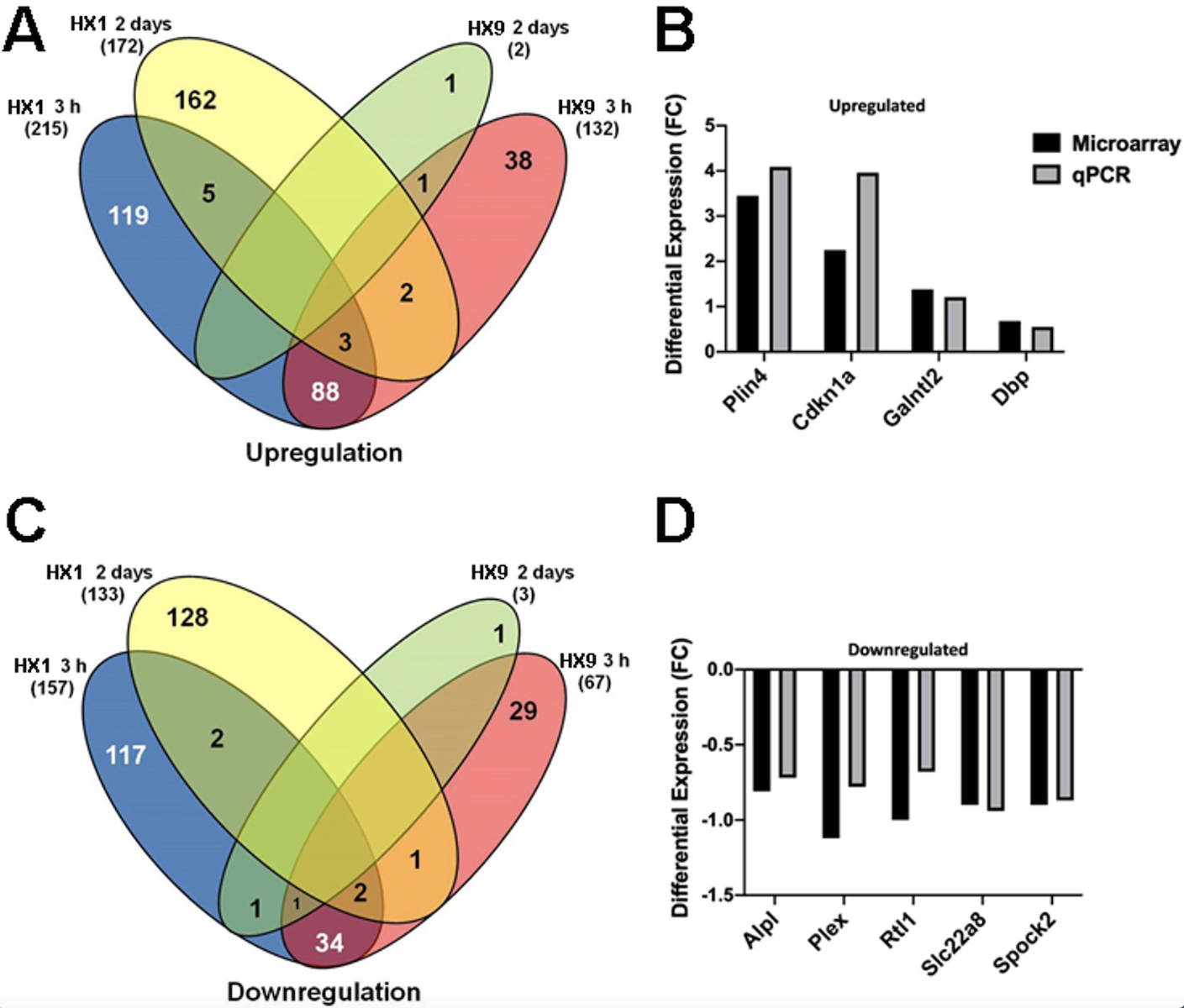
In a new window | Download PPT
Figure 1: Gene clustering and validation of microarray dataset by qPCR. (A, C) There is minimal commonality in the robust gene expression occurring at 3 h and 2 days following a single hypoxic exposure (HX1, left side) or 9 hypoxic exposures (HX9, right side) for either (A) upregulated or (C) downregulated gene expression. (B, D) Prior to bioinformatic analysis, qPCR was performed on several differentially expressed upregulated (B) and downregulated (D) genes for validation. Shown is the differential expression of analyzed genes, by fold-change (FC), from samples obtained from the naïve (i.e., untreated) experimental group.
Our qPCR analysis of expression changes of a selection of nine top-ranking (by fold-change) upregulated and downregulated genes identified in our microarray analysis revealed generally strong agreement between the two methods regarding the extent of differential expression, which served to validate the microarray methodology (Figure 1B, D).
Figure 2 highlights the “top 5” of several predictive bioinformatics-generated categories, and the “top 5” genes actually detected by microarray, for each respective experimental condition. To amplify differences amongst these groups, we compared the respective Biological Functions, Upstream Regulators, and Canonical Pathways they shared using a 'Comparison Analysis' (Figure 3), which also revealed the overall directional changes (up- or down-regulation) for each.
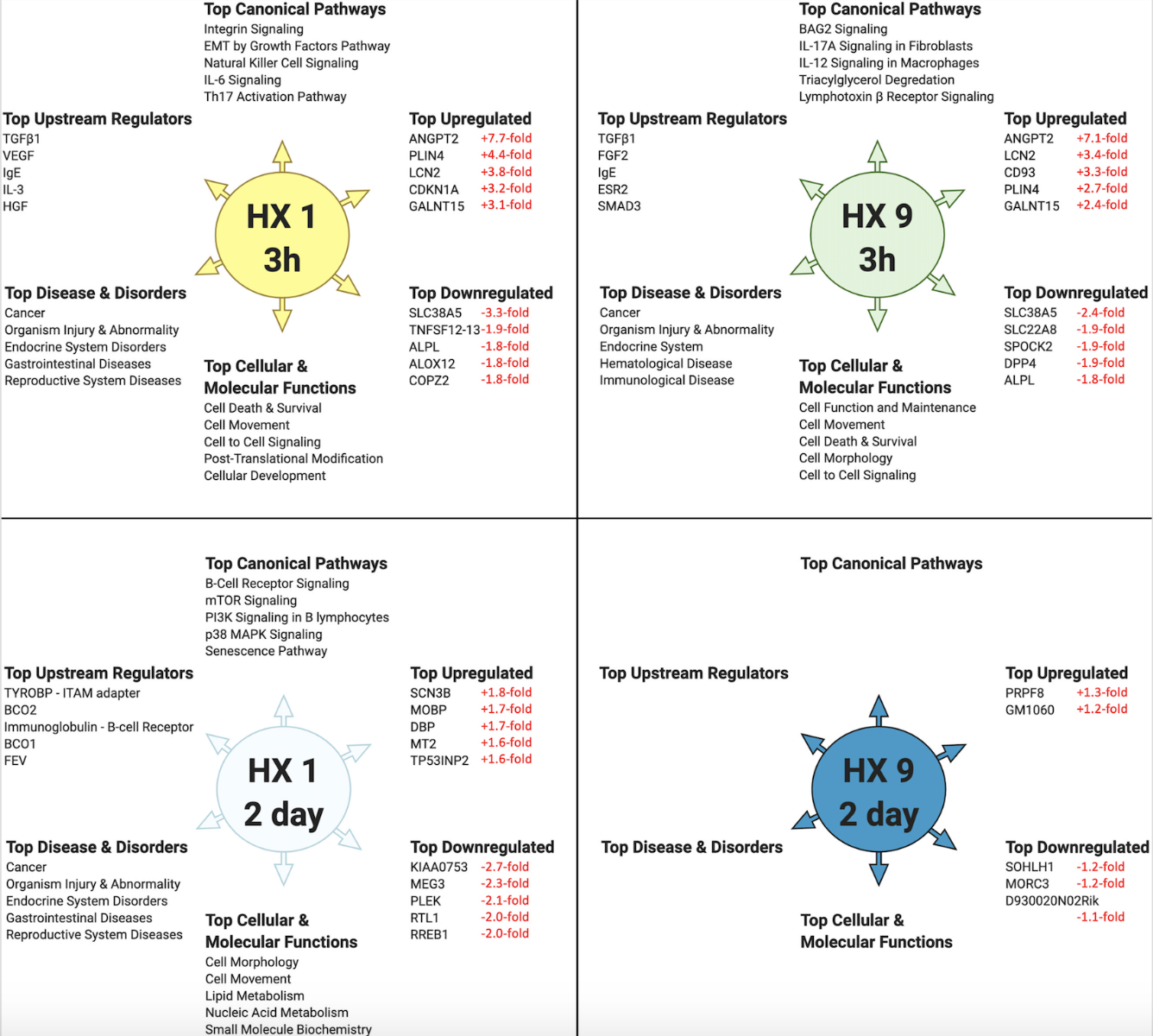
In a new window | Download PPT
Figure 2: Top-ranked bioinformatic findings based on the differentially expressed cerebral microvessel genes for each experimental preconditioning protocol and time point, relative to untreated naïve mice. IPA’s ‘Core Analyses’ were performed on microvessel microarray results at two time points following HX1 or HX9 conditioning. Shown (counterclockwise from top) are the top-ranked Canonical Pathways, Upstream Regulators, Diseases & Disorders, and Cellular & Molecular Functions predicted for each experimental group from such analyses, as well as the top-ranked differentially downregulated and upregulated genes actually measured by microarray in that group (with respective fold-changes shown in red). Only Canonicals with a ‘z-score’ of greater than ± 2.0 (= 95% confidence interval) are shown.
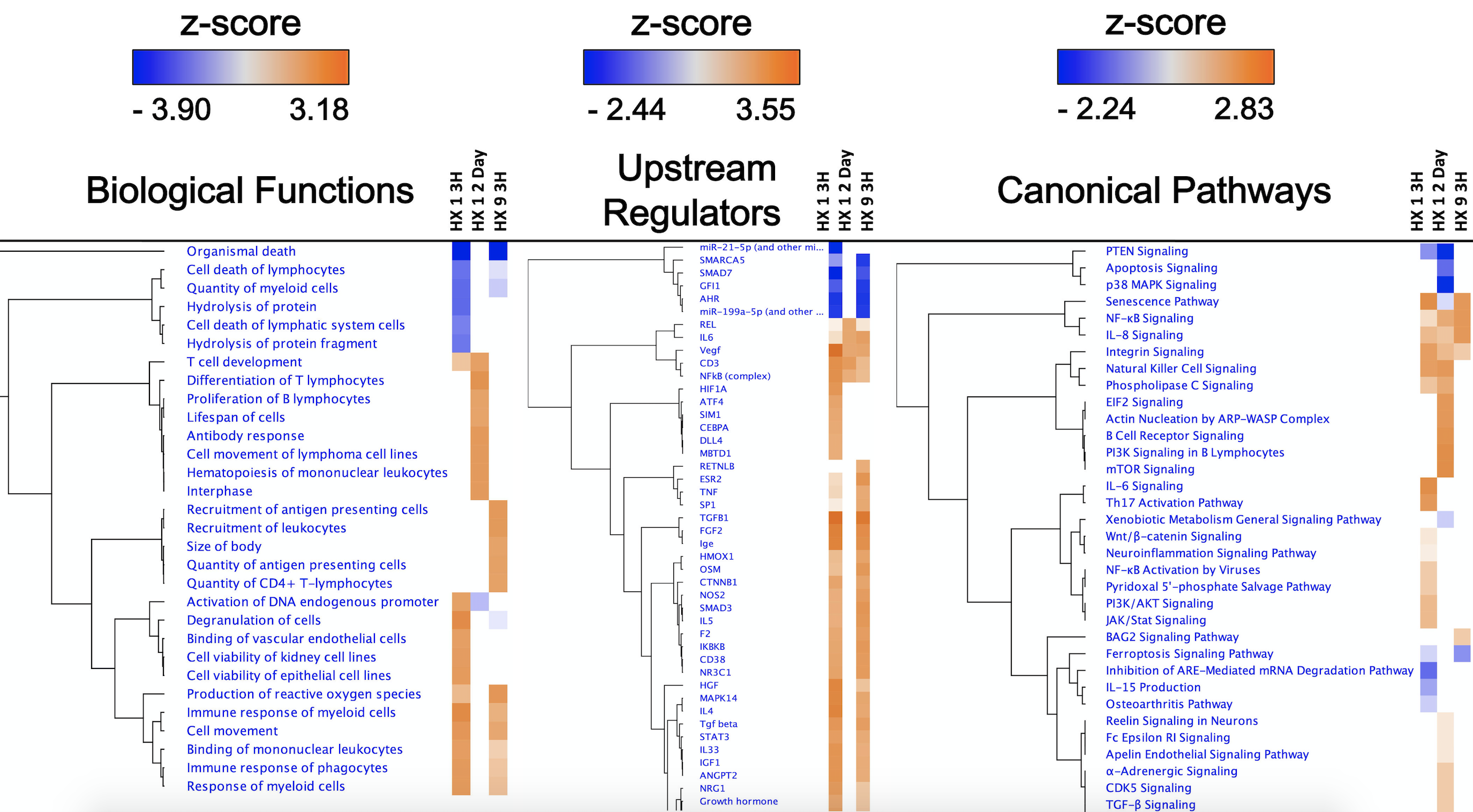
In a new window | Download PPT
Figure 3: Top-ranked Biological Functions, Upstream Regulators, and Canonical Pathways resulting from IPA’s ‘Comparison Analyses’ of the experimental groups. ‘Core Analyses’ performed in IPA for each of the three individual experimental groups (Fig. 2) were subsequently compared with each other in IPA’s ‘Comparison Analyses’ function to yield the most enriched Biological Functions (left), Upstream Regulators (center), and Canonical Pathways (right) that are shared among the groups. The respective z-score-based heat maps for each Function, Regulator, and Canonical are shown for each experimental group, with orange and blue color intensities representing the z-score-based extent of activation or inhibition, respectively, of a given Function/Regulator/Pathway, for that experimental group. Data were filtered by z-score (z-score > ± 2.0), followed by hierarchical clustering.
Generally speaking, our results were not necessarily predictable, at least as far as prototypical hypoxic signaling is concerned. By employing an unbiased, genome-wide analysis, we revealed insights into novel signaling changes that may be critical to promoting the temporally-unique ischemia-resilient phenotypes induced by HX1 and HX9. Conversely, we found that hypoxia-inducible factor (HIF) and other well-described hypoxia-regulated transcription factors (e.g., nuclear factor erythroid 2-related factor 2 (Nrf2), cAMP-response element binding protein (CREB), activator protein 1 (AP-1), early growth response protein 1 (Egr-1), heme oxygenase-1, and signal transducer and activator of transcription (STAT)), as well as other more well-known transcriptional and translational regulators of hypoxic responses (e.g., Rho GTPases, AMP-activated protein kinase (AMPK), reactive oxygen species (ROS), protein kinase PERK, Akt and other kinases (PERK), cyclooxygenase, and miRNAs) did not exhibit significant changes in expression in our samples. However, the time points at which we performed our microvessel expression analyses likely influenced these outcomes: Whether exposed to one or nine “doses” of hypoxia, microvessel samples were obtained 3 h or 2 days after the return to normoxic conditions, and not during the hypoxic exposure itself; thus, we may have missed transient changes in the expression of some of these molecular regulators (Thomas & Ashcroft, 2019). Regional differences in the hypoxic response may also underlie what might be considered unexpected findings (Xu et al., 2011). Moreover, the regulation of gene expression in response to hypoxia is, like many other stimuli, dependent on the intensity, duration, on/off rate, and frequency of the epigenetic stimulus, with the complexity of the cellular response not likely defined in any kind of one-size-fits-all manner.
Our bioinformatic analyses led us to advance the hypotheses that, independent of whether the hypoxic conditioning stimulus was single or repetitive, early activation of inflammatory and immune signaling pathways in microvessels that – true to Paracelsus “the dose makes the poison” adage – are not injurious but rather are integral to setting in motion the adaptive response. This is evident from our bioinformatics findings for the HX1-3h and HX9-3h groups with respect to the Canonical Pathways, Biological Functions, and Upstream Regulators that were enriched in genes differentially expressed by these two groups, relative to the HX1-2 day group (Figure 3). The left side panel of Figure 4 summarizes some of these predicted early signaling networks. Of note, nuclear factor kappa B (NFκB) activation secondary to ROS and inflammatory interleukin (IL) signaling, and the activation of Janus-faced signaling pathways involving transforming growth factor (TGF)-β and p38-mitogen-activated protein kinase (MAPK), are implicated in playing prominent roles in the initial hours after hypoxic conditioning, but then become inhibited and give way to the delayed expression of other mediators that transiently lock in the tolerant phenotype 24-72 hours later (reflected by the 2-day post-HX transcriptome). The latter likely occurs, in part, by reductions in the expression of IRAK2 (which encodes for the IL 1-receptor associated kinase and was downregulated 1.3-fold in the HX1-2day group), which in turn suppresses NFκB-driven transcriptional responses.
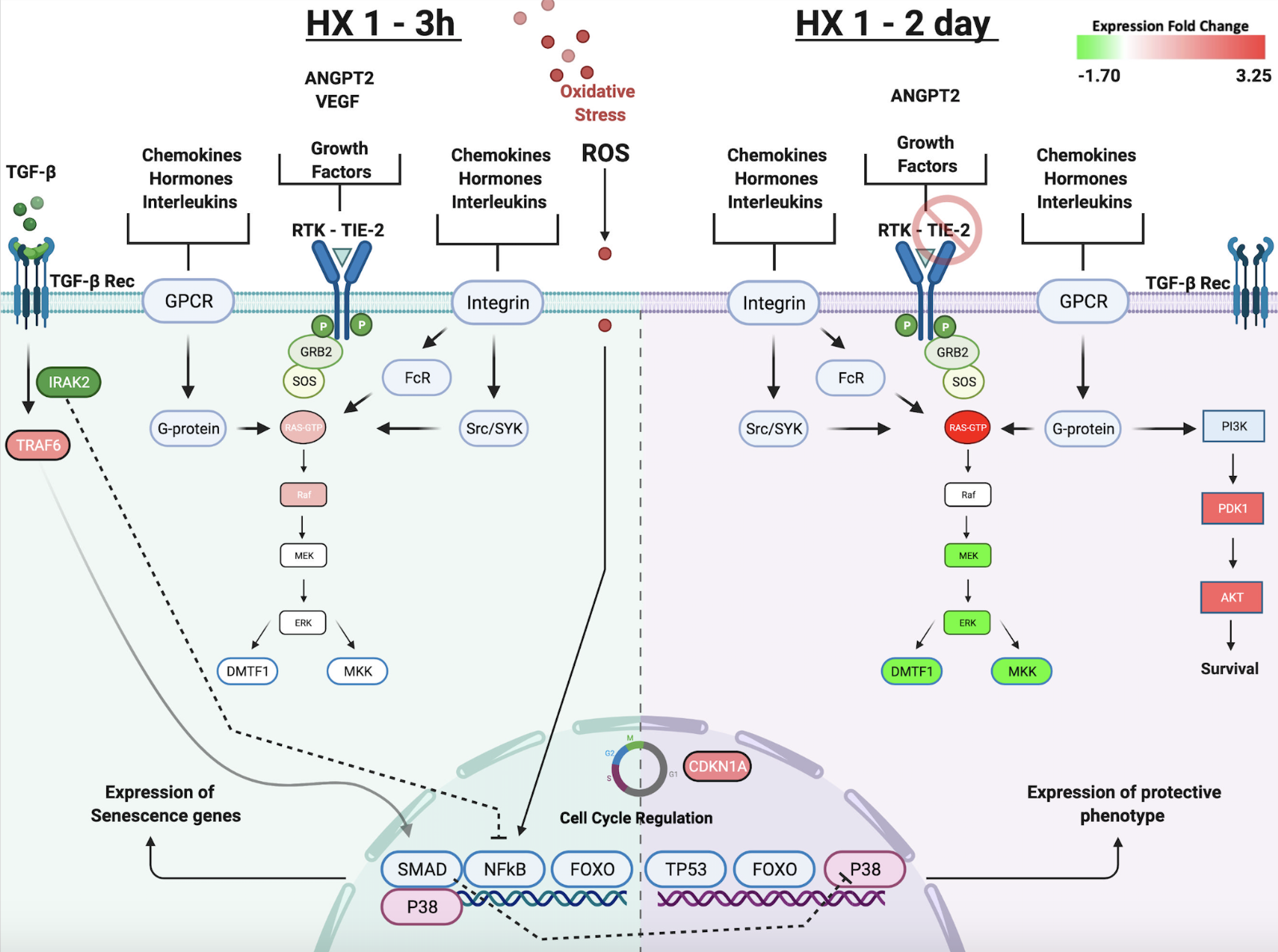
In a new window | Download PPT
Figure 4: The temporal response to HX1 conditioning. Left panel highlights signal transduction events occurring 3 h after HX1 that our microarray and bioinformatic analyses suggest contribute to ischemic tolerance observed two days later. Hypoxia-induced increases in ROS and other inflammatory mediators activate ANGPT2–TIE-2 signaling, and extensive integrin and receptor tyrosine kinase (RTK) receptor signaling, that is held in check by TGFβ. Downstream p38-MAPK activity is critical to regulating gene expression changes at this time. Overall, signaling in the senescence pathway is increased, promoting cell cycle arrest. Right panel highlights differential gene expression two days after HX1, concomitant with ischemic tolerance. With TIE-2 expression no longer reduced at this time, distinct effects of increased ANGPT2 expression are induced relative to those occurring 3 h after HX1. Increases in Ras signaling are nullified by reductions in the expression of its downstream effectors, which, together with now quiescent TGFβ receptor-mediated signaling, no longer drive p38-MAPK. Reductions in DMTF1 indirectly promote TP53 signaling by negatively regulating p19 and MDM2 (not shown). Together, increased TP53 and CDKN1A activity invoke changes in cell cycle regulation. Increases in PI3K signaling activate Akt-mediated survival pathways. This tolerant phenotype is associated with a downregulation of senescence pathway signaling.
Specifically, HX-induced increases in ROS and inflammatory mediators trigger TGF-β receptor activation (Figure 4), as well as increases in the E3 ubiquitin ligase tumor necrosis factor receptor-associated factor 6 (TRAF6), which together can upregulate transcriptional activity through myocyte enhancer factor 2 (MEF2) and Elk1, increase protein synthesis and related anabolic pathways, and reduce apoptosis-activating pathways secondary to phosphorylation of the pro-death transcription factors apoptosis signal-regulated kinase 1 (ASK1) and TGF-β activated kinase 1 (TAK1) (based on z-scores). The Smad superfamily members (Liu and Desai, 2015), as well as MAPK proteins, are integral to the transduction of TGF-β receptor-mediated signaling. The acute activation of cytoplasmic Smad 3 and 4, which facilitates TGF-β signaling, and the acute inhibition of nuclear Smad 7, which in turn suppresses TGF-β signaling, were predicted early after HX1 and HX9, but only transiently, as the expression levels of all three were unchanged two days later. TRAF6 was also downregulated two days after HX1, which would also help suppress TGF-β – NFκB signaling at this time.
The involvement of nine MAPK isoforms (all with significant p-values-of-overlap) was predicted by our bioinformatic analyses across all three experimental groups. Some were shared (MAP3K8), some (MAP3K1) were unique to HX1-3h, while others (MAPK, MAPK2K6 and MAP3K14) were unique to HX9-3h, with a significant z-score giving high confidence to the participation of MAP3K14, known more commonly as p38-MAPK, in HX9 signaling (Figure 4). The 1.3-fold increase in the expression of the MAP3K8 isoform 3 h after both HX1 and HX9, together with 1.3- and 1.2-fold increases in TRAF6 under these same conditions, likely mediate CD40-dependent signaling to activate extracellular signal-regulated kinase (ERK) in B cells and macrophages. Conversely, the MAPK isoform profile two days after HX1 conditioning was unique, with 1.3-fold reductions in the MAP2K6 and MAPKAPK5 isoforms, and a 1.5-fold increase in the MAP4K3 isoform. Thus, distinct activation/suppression profiles for MAPK isoform family members likely reflect, in turn, their distinct roles in early signal transduction responses to single or repetitive hypoxic conditioning, which may, in turn, explain the duration of their resultant ischemia-tolerant phenotypes.
Many of the hypoxia-induced pathways implicated, based on z-scores, in our bioinformatic analyses also involve signaling by receptor tyrosine kinases (RTKs) (Figures 4 and 5). RTKs are the second largest family of membrane receptors; working with membrane adaptors that in turn bind cytoplasmic substrates, RTKs activate downstream signaling pathways that involve phosphoinositide 3-kinase (PI3K), MAPK, and protein kinse c (PKC) intermediates (Paul and Hristova, 2019). Of note, TIE2 is an endothelial cell-specific RTK receptor that responds to a variety of ligands, including angiopoietin 1 (ANGPT1) and vascular endothelial growth factor (VEGF). ANGPT2 exerts a variety of context-dependent effects on endothelial cells. It negatively regulates the pro-angiogenic effects of ANGPT1 (Korhonen et al., 2016) as a result of its TEC-mediated antagonism of the TIE2 receptor, and it also controls the responsiveness of endothelial cells to exogenous cytokines (Felcht et al., 2012), thus regulating vascular remodeling and immune interactions driven by growth factors and inflammatory cytokines (Korhonen et al., 2016) produced either systemically or by CNS cell populations affected by hypoxia. Specifically, 3 h after both HX1 and HX9 conditioning, ANGPT2 was robustly (~7-fold) upregulated, as were many of its upstream regulators (e.g., VEGF, fibroblast growth factor 2 (FGF2), delta-like ligand 4 (DLL4), and insulin growth factor 1 (IGF1)). Interestingly, TIE2 receptor expression was decreased 1.6-fold 3 h after HX1, whereas it was unchanged 3 h after HX9. As a result of ANGPT2-mediated activation of the TIE2 receptor (by phosphorylation), leukocyte recruitment, chemotaxis, and degranulation are likely promoted, secondary to PI3K- and pyruvate dehydrogenase kinase 1 (PDK1)-mediated phosphorylation of Akt (Figure 4) (Korhonen et al., 2016). The same ANGPT2-TIE2 ligand-receptor interaction would also activate monocyte and integrin α5β1–mediated macrophage activation, recruitment, and infiltration (Scholz et al., 2011). While the exact mechanisms are unclear, we speculate that differences in ANGPT2-TIE2 signaling in HX1 and HX9 may serve as a pivotal biological switch (Thamm and David, 2016) for regulating downstream inflammatory signaling (Figure 6C), and thus may be one potential mechanism by which different durations of ischemic tolerance induced by HX1 and HX9 are ultimately established. In contrast, when the cerebral microvasculature was temporally defined by an ischemia-tolerant phenotype (2 days after HX1 and HX9), both ANGPT2 and TIE2 expression had returned to baseline.
As a result of the hypoxia-driven activation of p38-MAPK signaling secondary to a ROS-mediated activation of ERK (Di Micco et al., 2006), NFκb signaling secondary to tumor necrosis factor (TNF) α, and tumor protein 53 (TP53) signaling, a senescence-associated secretory phenotype (SASP) is induced wherein cells are irreversibly arrested from proliferating (Xu et al., 2014; Nakao et al., 2020). This process is maintained by cytokines like IL-1, and indirectly by cyclin-dependent kinase inhibitor 2A (CNDKN2A)-PRB (Chien et al., 2011; Anerillas et al., 2020), which inhibits Rb phosphorylation to halt the cell cycle. ‘Cell Senescence’ was the single Canonical pathway that exhibited an “anti” expression profile between the 3-h and 2-day comparisons (Figure 3), based on both protein fold-change and bioinformatic z-scores. In Figure 4, we re-created (with reduced complexity) several signaling pathways that were predicted in these datasets and illustrate how cell cycle regulation can be modulated through RTK signaling initiated early (3h) after HX1 and HX9. Two days later, MEK, ERK, MKK, and cyclin-D-binding Myb-like transcription factor 1 (DMTF1) are suppressed, resulting in CDKN1A upregulation, an increase in TP53 activity, and an overall reduction in senescence pathway activity.
Along with the aforementioned RTK receptors, integrin receptors also exhibited, based on z-score, the most enriched receptor-mediated signaling pathways identified in our datasets – particularly the acute responses to HX1 and HX9 (Figure 5). Integrin receptors, showing lower binding affinity than TIE2 (Felcht et al., 2012), were also likely activated by the aforementioned robust, HX1- and HX9-induced increases in ANGPT2 expression, consistent with the predicted upregulation of T cell activity at the 3-h signaling time points, and the predicted upregulation of B cell activity at the 2-day tolerance time point (Figure 5B and 5C, respectively). Through immunoreceptor tyrosine-based activation motifs (ITAMs) on Fc receptors, immune signaling activates Src-family tyrosine kinases and spleen tyrosine kinase (SYK)-family tyrosine kinases that connect transduced signals to common survival pathways shared with other receptors, as evidenced by the SYK-mediated phosphorylation and activation of the SH2 adapter proteins bruton tyrosine kinase (BTK, 1.2-fold increase, and significant z-score [2.0] for HX9-3h group) and phospholipase C (PLC, z-score = 1.7 for the HX1-2d group). Both BTK and PLC were also upregulated in ischemia-tolerant (HX1-2d) microvasculature. Alpha-integrin receptor subunit/ITAM signaling, through SYK, also drives T cell (Figure 5, panel B) and B cell (Figure 5, panel C) activation (Barb, 2020). In fact, the T-cell adapter CD28 (Figure 5, panel B) was one of the top-ranked upstream regulators.

In a new window | Download PPT
Figure 5: An integrated synthesis of potential signal transduction responses to repetitive (HX9) preconditioning. A) Bioinformatic analyses revealed that preconditioning modulates receptor-mediated signaling activity through: Integrin receptors via SYK activation (left); RTK receptor signaling (such as VEGF, ERBB [estrogen-like], and growth factor receptors (center); and EGFRs and solute carrier complexes (SLCs) that suppress ferroptosis by regulating TP53 activity. Overall, these signaling pathways result in the expression of genes promoting cell survival (including those inhibiting ferroptosis), as well as the expression of pro-inflammatory and immune genes (see Figs. 6 and 7). The former is more robust after HX9, the latter more dampened, relative to the expression changes triggered by HX1. B) and C) provide details for the activation and signaling of T cell receptors (TCR) and B cell receptors (BCR) shown in the red box in Panel A. Panel B shows the consequences of T cell activation occurring 3 h after both HX1 and HX9. The surface protein CD28, which serves as a co-stimulus required for T-cell activation and survival, was a significant upstream regulator in both groups. T cells produce TGFβ, which drives early signaling (Fig. 4), IL-17 from its CD4+ subset (consistent with ‘Th17 Activation Pathway’ being a top Canonical), and IL-6, TNFα, interferon gamma (IFNγ), and other cytokines that our bioinformatic findings, and existing literature, support as mediators of conditioning-induced signal transduction. Panel C relates to BCR signaling, which was activated in a delayed fashion 2 days after HX1 (and was the top Canonical in this group). Of note, BCR signaling reduces apoptosis through PI3K/Akt, and drives activation of the NFAT transcription factor family through SYK.
ITAM-mediated signaling, which is coupled to α-integrin receptor subunits, can also lead to the formation of the CARD-BCL-MALT Signalosome Complex Assembly, comprised of the caspase-recruit domain (CARD) protein, the adapter proteins of B cell lymphomas (BCL), and the mucosa-associated lymphoid type para-caspase 1 (MALT1). BCL-10 and MALT1 are key regulators of NFκB as well as physiological antigen receptor signaling in B cells and T cells, which are required for adaptive immunity (Ruefli-Brasse et al., 2003; Mócsai et al., 2010; Mócsai et al., 2015; Juilland and Thome, 2018). Evidence for this signaling was identified in the ischemia-tolerant (HX1-2d and HX9-3h) microvasculature, with CARD8 (predicted with p < 0.04) and BCL6 (measured 1.2 fold-change, p < 0.002) enriched in the HX-2d group, while the HX9-3h group exhibited four BCL-type isoforms (BCL11B [predicted with p < 0.02]; BCL2L11 [predicted with p<0.02], BCL2L12 [predicted with p < 0.001], and BCL6 [inhibited, z-score = -1.4, with p < 0.0007]), which likely drove reductions in proinflammatory NFκB signaling.
Our microarray and bioinformatic findings are consistent with our previous studies showing that both HX1 and HX9 conditioning stimuli induce immunophenotypic changes in the innate and adaptive immune systems in the periphery, with immune cell recruitment into the tolerant brain regulated by the cerebral vasculature. Specifically, 12 h after HX1, B cell representation in blood was doubled, with concomitant reductions of monocytes and T cell populations (Stowe et al., 2012). As described above, our microvessel transcriptomics show an early HX1-3h integrin activation (Figure 5A) that likely drive this monocyte recruitment, as well as early activation of T-cell receptor (TCR) signaling (Figure 5B). TCR activation directly affects recruitment of T cells into the brain (Gaylo et al., 2016) and is predicted to be upregulated after HX9 (Figure 5B). Coupled with predicted increases in integrin signaling expression, these findings are all suggestive of T cell trafficking early after hypoxia. We also identified a delayed activation of B cell receptor (BCR) signaling in microvessels from the HX1-2d cohort (Figure 5C), at the time of brain tolerance to ischemia. Several B cell subsets are both anti-inflammatory and neurotrophic (Selvaraj et al., 2016). Thus, B cell activation may be a conserved neuroprotective mechanism similar to other forms of short-term tolerant phenotypes, including those induced by voluntary exercise conditioning, which we also found transcriptionally activates B cells through the upregulation of BCR signaling (Poinsatte et al., 2019).
Brain microvessel recruitment of B cells, but not T cells, is heightened after HX9 (Monson et al., 2014; Selvaraj et al., 2017), with tolerant microvessels upregulating C-X-C motif chemokine ligand 13 (CXCL13), the chemokine that specifically recruits B cells, 24 h earlier than microvessels isolated from non-conditioned animals following transient focal stroke (Monson et al., 2014). Additional neuroinflammatory signaling 3 h after HX9 includes IL-8, IL-12, IL-15, and IL-17 (Figure 7), although this signaling completely declines by two days. While B cells are activated in the periphery by HX1, peripheral populations shift to an anti-inflammatory phenotype in response to the subsequent hypoxic exposures that define HX9, with antibody production and antigen recognition progressively downregulated (Monson et al., 2014), in parallel to the downregulation of selectins and integrins within the HX9-treated microvessels after stroke (Stowe et al., 2011). Taken together, these findings provide insights into the complex and coordinated interplay between the cerebral endothelium and circulating immune cell populations triggered by hypoxic conditioning that ultimately mitigate ischemic injury and neuroinflammation.

In a new window | Download PPT
Figure 7: 'Comparison Analyses’ heatmaps of immunoregulatory genes in the three preconditioned experimental groups. ‘Core Analyses’ were performed in IPA for each respective experimental group (see Fig. 2) and were then subsequently compared with each other using a ‘Comparison Analyses’ function (see Figure 3) to yield the most enriched Canonical Pathways related to immunity and inflammation that are shared among the groups. Heatmaps of differentially expressed genes were created for each respective signaling pathway; red indicates activation/upregulation of the gene, green indicates suppression/downregulation of the gene, grey indicates changes that fell below our threshold cutoff (1.3 fold-change), and white indicates a gene was not detected, for each respective reference being compared across experimental conditions, in fold-change units, per the scale shown (top right).
Ultimately, cell death signaling, and effector pathways are likely suppressed to achieve neurovascular protection by hypoxic and other forms of conditioning. Ferroptosis is a non-apoptotic form of cell death activated by the iron-dependent accumulation of lipid peroxides and depletion of plasma membrane polyunsaturated fatty acids (Cao and Dixon, 2016) secondary to conditions that inhibit glutathione biosynthesis or the glutathione-dependent antioxidant enzyme glutathione peroxidase 4 (GPX4), or by cysteine starvation (Kang et al., 2021). This death-signaling pathway was predicted to be downregulated acutely following HX9, and also to a lesser extent following HX1, which may result from a hypoxic conditioning-mediated upregulation of the ferroptosis suppressor CDK1NA. (Figure 5) and upregulation of Nrf2 signaling (Kang et al., 2021).
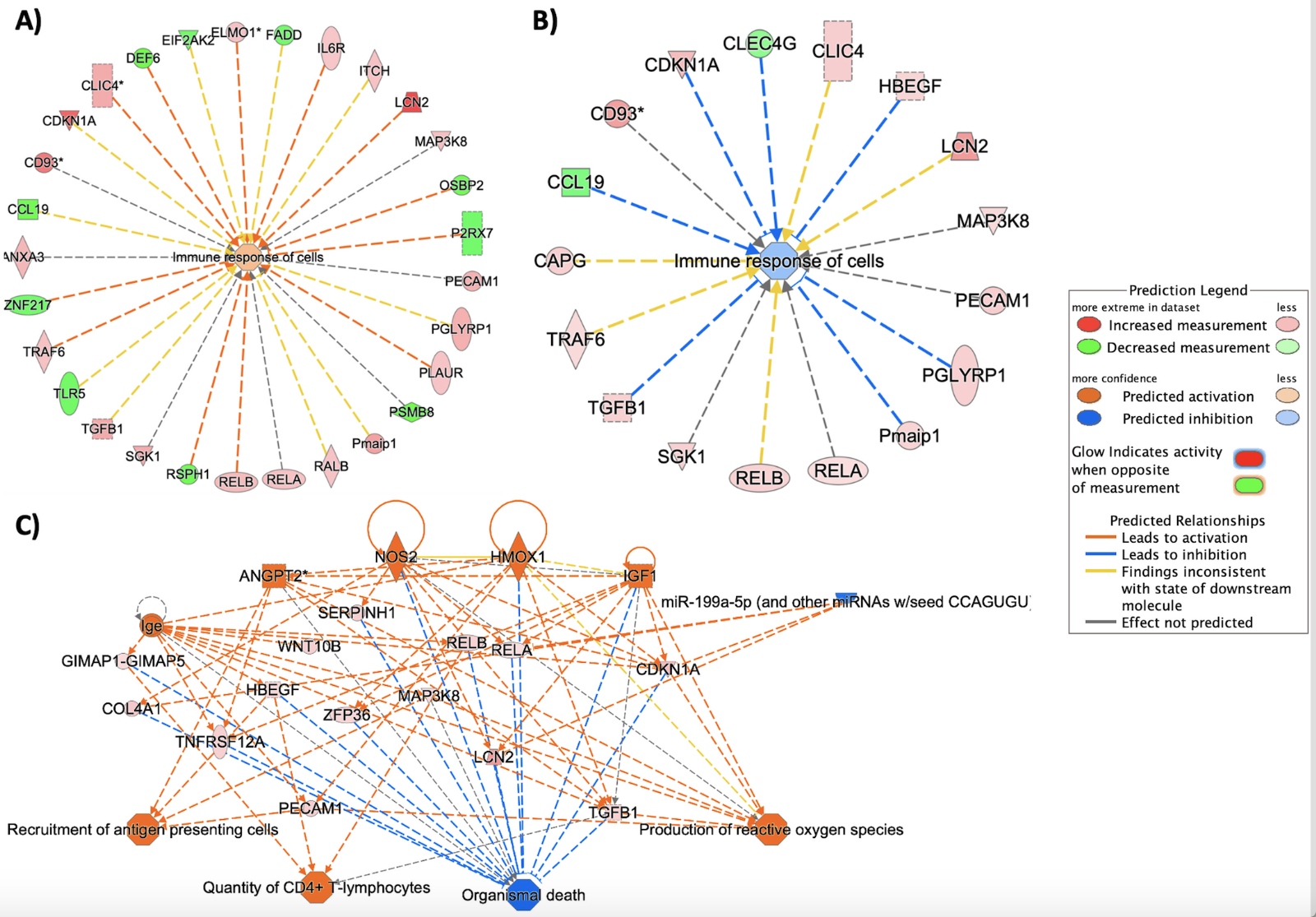
Six-transmembrane epithelial antigen of prostate 3 (STEAP3) encodes a multi-pass membrane iron and copper transporter that regulates heme homeostasis; because it was identified as one of the top-ranking upstream regulators exclusive to the HX9-3h experimental group (ranked by p-value-of-overlap < 0.0001), and ‘Ferroptosis’ was one of the few downregulated canonical pathways in this group, we hypothesize heme signaling may participate in driving long-lasting periods of tolerance in response to repetitive conditioning. Moreover, STEAP3 is a direct target of p53, and works with p53 to regulate exosome secretions (Wu et al., 2020). As mentioned earlier, p53 may be inhibiting active dipeptidyl peptidase (DPP4) and/or upregulating CDKN1A (Figure 5B) to regulate cellular senescence (Figure 4), and through decreasing lipid peroxidation and NADPH oxidase (NOX)-driven ROS production may suppress pro-inflammatory lipid signaling and ferroptosis. The 3.8- and 3.4-fold upregulation of astrocyte-derived (Ranjbar et al., 2019) lipocalin 2 (LCN2) in HX1-3h (Figure 6A) and HX9-3h (Figure 6B) groups also supports heme-based signaling mechanisms, as well as the immune response of cells (Figure 6C).
In a new window | Download PPT
Figure 6: Repetitive preconditioning (HX9) suppresses cell death/apoptosis through regulation of the innate immune response. A), B) This two-part figure represents the bioinformatic enrichment of immune-type proteins and the differential expression of implicated proteins. Proteins that were differentially expressed and classified in IPA’s “Immune response of cells” Biological Function category are displayed above for the A) HX1-3h timepoint, and B) the HX9-3h timepoint. Actual microarray expression data and predictive analysis features were both applied to generate this graphic. Orange nodes represent ‘activated’ or upregulated activity while blue nodes represent a suppression or down regulation, each with z-scores (>2). Nodes that are red were genes upregulated in the microarray dataset while green nodes indicate a down regulation. Connecting lines in yellow indicate upstream or downstream activity in our dataset that is inconsistent with the state of downstream molecule in the IPA database, suggesting an atypical activity of the particular protein-protein relation shown, potentially reflective of unique effects of our preconditioning treatments. C) IPA-generated ‘Mechanistic Network’ reveals protein-protein interactions with downstream cellular functions for HX9-preconditioned microvessels. With the exception of the single miRNA (blue), all of the protein interactions above indicate increasing protein activity or expression (indicated by orange and red nodes) in a way that ultimately results in reduction of organismal death (as indicated by converging blue lines).
The solute carrier SLC38A5 (which functions as an amino acid and anion transporter) was the most downregulated gene 3 h after both HX1 (-3.3-fold) and HX9 (-2.4-fold). Unique to the HX9-3h group, SLC22A8 (downregulated 1.9-fold) was the 2nd-most downregulated gene. SLC22A8 functions to excrete and detoxify organic anions (Sykes et al., 2004) and mediate the uptake of the proinflammatory prostaglandins E2 and F2α (Sweet et al., 2002). Given the 1.7-fold suppression of arachidonic acid lipoxygenase (ALOX12) in the ischemia-tolerant (HX9-3h) microvasculature, we speculate that SLC22A8 reduces the proinflammatory lipid peroxidation that drives ferroptosis. In sum, HX9 results in myriad changes in microvessel gene expression related to the suppression of lipid peroxidation and ferroptosis, which, if sustained, may also contribute to the long-lasting tolerant phenotype it induces.
Study Limitations. Our study was limited by analyzing cerebral microvessels from only one sex (males); given that recent investigations from our lab and others have identified sexual dimorphism at the proteome level in the brain microcirculation (Cikic et al., 2020), these differences should be assessed in future studies. Also, the differential centrifugation method used to obtain the microvessel-rich preparations we analyzed yielded endothelial cells from arterioles, capillaries, and venules, along with arteriolar and venular smooth muscle. Thus, our results represent an amalgam of the well-described, significant heterogeneity of the microcirculation (Chi et al., 2003; Macdonald et al., 2010). We utilized qPCR for validating an assortment of genes exhibiting significantly changed expression, but did not perform mRNA- or protein-level validation of key molecules in the pathways predicted by our bioinformatic analyses to be activated or inhibited. A proteomics-based study would have brought us closer to understanding the months-long stroke-tolerant phenotype established by HX9, given the relative quiescence of the microvessel transcriptome just days after the stimulus. Nevertheless, for the first time, we have identified time-dependent signaling and activation pathways within microvessels that are uniquely dependent on the number of systemic conditioning stimuli. Several of these pathways could be activated/suppressed to establish the anti-inflammatory and neurovascular-protective phenotypes that define ischemic tolerance and could therefore serve as therapeutic targets to reduce the devastating effects of cerebral ischemia on brain health and function.
Acknowledgments
Supported by NIH R01 HL79278 (JMG), NIH R01 NS088555 (AMS), American Heart Association (AMS), and The Hope Center for Neurological Disorders at Washington University School of Medicine. We thank Jennifer Perfater and Angie B. Freie for expert technical assistance with this study, and Seth Crosby, MD for his valuable direction with the microarray studies and early statistical analyses. We also thank the following content creators at www.Biorender.com: Anna Lazaratos – for Panel B of Figure 5 (taken from template - T cell activation and differentiation), Audrey Kassardjian, Jean-Philippe Julien, and Taylor Sicard – for panel C of figure 5 (adapted from template – Targeting B cell co-receptors for vaccine design).
Authors contributions
AMS and JMG conceived the study; AMS and JR designed the PCR and microarray experiments; JCH and DO performed the bioinformatic analysis; JCH, DO, AMS, and JMG interpreted experimental and bioinformatic results; JCH prepared figures; and JCH, JMG, and AMS drafted, edited, and made final revisions to the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration of competing interests
The authors declare no competing interests.
References
Jarrod C. Harman1,2,3
1Departments of Ophthalmology, 2Biochemistry & Molecular Biology, 3Neuroscience Center of Excellence, Louisiana State University School of Medicine, LSUHSC, New Orleans, LA
David A. Otohinoyi2
2Departments of Biochemistry & Molecular Biology, Louisiana State University School of Medicine, LSUHSC, New Orleans, LA
John W. Reitnauer III4
Department of Neurology, University of Kentucky College of Medicine, Lexington, KY
Ann M. Stowe4
Department of Neurology, University of Kentucky College of Medicine, Lexington, KY
Jeff M. Gidday1,2,3,5
1Departments of Ophthalmology, 2Biochemistry & Molecular Biology, 3Neuroscience Center of Excellence, and 5Physiology, Louisiana State University School of Medicine, LSUHSC, New Orleans, LA
Corresponding author:
Jeff Gidday
Email: jgidda@lsuhsc.edu
or
Jarrod Harman
Email: jharma@lsuhsc.edu
In a new window | Download PPT
Figure 1: Gene clustering and validation of microarray dataset by qPCR. (A, C) There is minimal commonality in the robust gene expression occurring at 3 h and 2 days following a single hypoxic exposure (HX1, left side) or 9 hypoxic exposures (HX9, right side) for either (A) upregulated or (C) downregulated gene expression. (B, D) Prior to bioinformatic analysis, qPCR was performed on several differentially expressed upregulated (B) and downregulated (D) genes for validation. Shown is the differential expression of analyzed genes, by fold-change (FC), from samples obtained from the naïve (i.e., untreated) experimental group.
In a new window | Download PPT
Figure 2: Top-ranked bioinformatic findings based on the differentially expressed cerebral microvessel genes for each experimental preconditioning protocol and time point, relative to untreated naïve mice. IPA’s ‘Core Analyses’ were performed on microvessel microarray results at two time points following HX1 or HX9 conditioning. Shown (counterclockwise from top) are the top-ranked Canonical Pathways, Upstream Regulators, Diseases & Disorders, and Cellular & Molecular Functions predicted for each experimental group from such analyses, as well as the top-ranked differentially downregulated and upregulated genes actually measured by microarray in that group (with respective fold-changes shown in red). Only Canonicals with a ‘z-score’ of greater than ± 2.0 (= 95% confidence interval) are shown.
In a new window | Download PPT
Figure 3: Top-ranked Biological Functions, Upstream Regulators, and Canonical Pathways resulting from IPA’s ‘Comparison Analyses’ of the experimental groups. ‘Core Analyses’ performed in IPA for each of the three individual experimental groups (Fig. 2) were subsequently compared with each other in IPA’s ‘Comparison Analyses’ function to yield the most enriched Biological Functions (left), Upstream Regulators (center), and Canonical Pathways (right) that are shared among the groups. The respective z-score-based heat maps for each Function, Regulator, and Canonical are shown for each experimental group, with orange and blue color intensities representing the z-score-based extent of activation or inhibition, respectively, of a given Function/Regulator/Pathway, for that experimental group. Data were filtered by z-score (z-score > ± 2.0), followed by hierarchical clustering.
In a new window | Download PPT
Figure 4: The temporal response to HX1 conditioning. Left panel highlights signal transduction events occurring 3 h after HX1 that our microarray and bioinformatic analyses suggest contribute to ischemic tolerance observed two days later. Hypoxia-induced increases in ROS and other inflammatory mediators activate ANGPT2–TIE-2 signaling, and extensive integrin and receptor tyrosine kinase (RTK) receptor signaling, that is held in check by TGFβ. Downstream p38-MAPK activity is critical to regulating gene expression changes at this time. Overall, signaling in the senescence pathway is increased, promoting cell cycle arrest. Right panel highlights differential gene expression two days after HX1, concomitant with ischemic tolerance. With TIE-2 expression no longer reduced at this time, distinct effects of increased ANGPT2 expression are induced relative to those occurring 3 h after HX1. Increases in Ras signaling are nullified by reductions in the expression of its downstream effectors, which, together with now quiescent TGFβ receptor-mediated signaling, no longer drive p38-MAPK. Reductions in DMTF1 indirectly promote TP53 signaling by negatively regulating p19 and MDM2 (not shown). Together, increased TP53 and CDKN1A activity invoke changes in cell cycle regulation. Increases in PI3K signaling activate Akt-mediated survival pathways. This tolerant phenotype is associated with a downregulation of senescence pathway signaling.
In a new window | Download PPT
Figure 5: An integrated synthesis of potential signal transduction responses to repetitive (HX9) preconditioning. A) Bioinformatic analyses revealed that preconditioning modulates receptor-mediated signaling activity through: Integrin receptors via SYK activation (left); RTK receptor signaling (such as VEGF, ERBB [estrogen-like], and growth factor receptors (center); and EGFRs and solute carrier complexes (SLCs) that suppress ferroptosis by regulating TP53 activity. Overall, these signaling pathways result in the expression of genes promoting cell survival (including those inhibiting ferroptosis), as well as the expression of pro-inflammatory and immune genes (see Figs. 6 and 7). The former is more robust after HX9, the latter more dampened, relative to the expression changes triggered by HX1. B) and C) provide details for the activation and signaling of T cell receptors (TCR) and B cell receptors (BCR) shown in the red box in Panel A. Panel B shows the consequences of T cell activation occurring 3 h after both HX1 and HX9. The surface protein CD28, which serves as a co-stimulus required for T-cell activation and survival, was a significant upstream regulator in both groups. T cells produce TGFβ, which drives early signaling (Fig. 4), IL-17 from its CD4+ subset (consistent with ‘Th17 Activation Pathway’ being a top Canonical), and IL-6, TNFα, interferon gamma (IFNγ), and other cytokines that our bioinformatic findings, and existing literature, support as mediators of conditioning-induced signal transduction. Panel C relates to BCR signaling, which was activated in a delayed fashion 2 days after HX1 (and was the top Canonical in this group). Of note, BCR signaling reduces apoptosis through PI3K/Akt, and drives activation of the NFAT transcription factor family through SYK.
In a new window | Download PPT
Figure 6: Repetitive preconditioning (HX9) suppresses cell death/apoptosis through regulation of the innate immune response. A), B) This two-part figure represents the bioinformatic enrichment of immune-type proteins and the differential expression of implicated proteins. Proteins that were differentially expressed and classified in IPA’s “Immune response of cells” Biological Function category are displayed above for the A) HX1-3h timepoint, and B) the HX9-3h timepoint. Actual microarray expression data and predictive analysis features were both applied to generate this graphic. Orange nodes represent ‘activated’ or upregulated activity while blue nodes represent a suppression or down regulation, each with z-scores (>2). Nodes that are red were genes upregulated in the microarray dataset while green nodes indicate a down regulation. Connecting lines in yellow indicate upstream or downstream activity in our dataset that is inconsistent with the state of downstream molecule in the IPA database, suggesting an atypical activity of the particular protein-protein relation shown, potentially reflective of unique effects of our preconditioning treatments. C) IPA-generated ‘Mechanistic Network’ reveals protein-protein interactions with downstream cellular functions for HX9-preconditioned microvessels. With the exception of the single miRNA (blue), all of the protein interactions above indicate increasing protein activity or expression (indicated by orange and red nodes) in a way that ultimately results in reduction of organismal death (as indicated by converging blue lines).
In a new window | Download PPT
Figure 7: 'Comparison Analyses’ heatmaps of immunoregulatory genes in the three preconditioned experimental groups. ‘Core Analyses’ were performed in IPA for each respective experimental group (see Fig. 2) and were then subsequently compared with each other using a ‘Comparison Analyses’ function (see Figure 3) to yield the most enriched Canonical Pathways related to immunity and inflammation that are shared among the groups. Heatmaps of differentially expressed genes were created for each respective signaling pathway; red indicates activation/upregulation of the gene, green indicates suppression/downregulation of the gene, grey indicates changes that fell below our threshold cutoff (1.3 fold-change), and white indicates a gene was not detected, for each respective reference being compared across experimental conditions, in fold-change units, per the scale shown (top right).
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 8109 | 14 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA