Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
STAT3 induces hypoxic preconditioning against oxidative stress in neural stem cells
Time:2024-03-08
Number:6018
Author Affiliations

Conditioning Medicine 2023. 6(4): 138-146.
Abstract
Although transplantation of neural stem cells (NSCs) has been proposed as a therapeutic strategy in stroke, oxidative stress in ischemic brains reduces the surviving transplanted cells. Hypoxic preconditioning (HP) improves the survivability of NSCs, but the mechanisms are obscure. Activation of signal transducer and activator of transcription 3 (STAT3) via Janus kinase 2 (JAK2) regulates proliferation in NSCs. However, the role of STAT3 in HP of NSCs has not been elucidated. This study investigated the role of STAT3 and representative downstream signals such as vascular endothelial growth factor (VEGF) and cyclin D1 in hypoxic preconditioned NSCs. NSCs were isolated from the subventricular zones of postnatal mice. NSCs were exposed to 5% hypoxia for 24 hours as HP and to H2O2 for 12 hours as lethal oxidative stress. HP decreased cell death and enhanced their viability against H2O2-mediated lethal oxidative stress. Western blot analysis revealed that phosphorylation of JAK2 and STAT3 was increased after HP. Moreover, HP increased the concentration of VEGF in the culture medium and the expression of cyclin D1 in NSCs. Administration of a STAT3 inhibitor suppressed the activation of STAT3 and the downstream signals, resulting in decreased efficacy of HP in oxidative injury. The activation of STAT3 may induce the HP effect in NSCs to protect against oxidative stress, and it could be a new therapeutic target to improve the efficacy of cell therapy in stroke.
Keywords: Hypoxic preconditioning, Neural stem cell, Oxidative stress, STAT3
Abstract
Although transplantation of neural stem cells (NSCs) has been proposed as a therapeutic strategy in stroke, oxidative stress in ischemic brains reduces the surviving transplanted cells. Hypoxic preconditioning (HP) improves the survivability of NSCs, but the mechanisms are obscure. Activation of signal transducer and activator of transcription 3 (STAT3) via Janus kinase 2 (JAK2) regulates proliferation in NSCs. However, the role of STAT3 in HP of NSCs has not been elucidated. This study investigated the role of STAT3 and representative downstream signals such as vascular endothelial growth factor (VEGF) and cyclin D1 in hypoxic preconditioned NSCs. NSCs were isolated from the subventricular zones of postnatal mice. NSCs were exposed to 5% hypoxia for 24 hours as HP and to H2O2 for 12 hours as lethal oxidative stress. HP decreased cell death and enhanced their viability against H2O2-mediated lethal oxidative stress. Western blot analysis revealed that phosphorylation of JAK2 and STAT3 was increased after HP. Moreover, HP increased the concentration of VEGF in the culture medium and the expression of cyclin D1 in NSCs. Administration of a STAT3 inhibitor suppressed the activation of STAT3 and the downstream signals, resulting in decreased efficacy of HP in oxidative injury. The activation of STAT3 may induce the HP effect in NSCs to protect against oxidative stress, and it could be a new therapeutic target to improve the efficacy of cell therapy in stroke.
Keywords: Hypoxic preconditioning, Neural stem cell, Oxidative stress, STAT3
Highlights
Post-stroke oxidative stress reduces transplanted neural stem cell (NSC) efficacy. Hypoxic preconditioning (HP) of NSCs may solve this, but the mechanisms are obscure. We studied JAK2-STAT3 pathway molecules in HP of mouse NSCs. HP induces oxidative stress tolerance in NSCs via the JAK2-STAT3 pathway. VEGF and cyclin D1 are downstream mediators of the acquisition of preconditioning.
Introduction
Transplantation of neural stem cells (NSCs) has been proposed as a promising therapeutic strategy in stroke. Previous studies have shown that transplantation of NSCs in the acute stage of both ischemic and hemorrhagic stroke reduces lesion size and inhibits apoptosis in the surrounding area by providing neuroprotective paracrine factors that enhance host cell survival and function (Bliss et al., 2007; Harms et al., 2010; Wakai et al., 2016). However, a hostile microenvironment does not permit the maintenance of high numbers of surviving transplanted cells, mainly due to the production of reactive oxygen species (Kinouchi et al., 1991; Savitz et al., 2002; Chen et al., 2011). This massive loss of stem cells post-engraftment is an impediment that lessens the effectiveness of cell transplantation therapy.
One approach for improving the ability of NSCs to survive in a harsh environment and enhancing their therapeutic efficacy is preconditioning the cells ex vivo in a hypoxic environment (Yu et al., 2013; Bernstock et al., 2017). This phenomenon, called hypoxic preconditioning (HP), activates endogenous defense mechanisms that provide marked protective effects against ischemic stroke and other acute attacks in the brain (Liu et al., 2000; Theus et al., 2008; Jaderstad et al., 2010; Wakai et al., 2016). In embryonic stem cells and mesenchymal stem cells, HP enhances proliferation, neuronal differentiation, paracrine activity leading to increased trophic support, and the capacity to home to the lesion site (Theus et al., 2008; Wang et al., 2008). Like many other cell types, NSCs acquire ischemic tolerance and regenerative properties after HP (Horie et al., 2008). Although some survival and protective molecules including hypoxia-inducible factor-1 (Yu et al., 2013), glycogen synthase kinase-3β, matrix metalloproteinase-2 (Li et al., 2008), survivin (Wisel et al., 2009), and Bcl-2 were reported to be involved in the response to HP, elucidation of the molecular mechanisms of HP in NSCs, which would provide new insight for improvement of cell therapy, has not been completely achieved.
Signal transducer and activator of transcription 3 (STAT3), which is a transcription factor as well as an intracellular signal transducer, is activated by various stimuli and molecules such as cytokines and growth factors, including interleukin-6, leukemia inhibitory factor, and ciliary neurotrophic factor (Darnell, 1997; Levy and Lee, 2002). The binding of these cytokines or growth factors to their cognate receptors induces phosphorylation and activation of Janus kinase (JAK) 2, which then activates STAT3 through phosphorylation of a tyrosine residue (Tyr 705) (Hebenstreit et al., 2005). Phosphorylated STAT3 forms dimers, translocates to the nucleus, binds to specific promoters of target genes, and induces expression of various genes such as vascular endothelial growth factor (VEGF) and cyclin D1 (Bromberg and Darnell, 2000). VEGF mainly has angiogenic effects, but paracrine VEGF also functions as a neuroprotective factor (Froger et al., 2020). On the other hand, cyclin D1 regulates the cell cycle and is involved in the proliferation of NSCs (Yang et al., 2016). Past reports have reported that STAT3 induces cell differentiation and regulates proliferation of NSCs (Yang et al., 2016; Cheng et al., 2019).
A role for STAT3 in the preconditioning induced by ischemia was initially reported by studies on sublethal ischemia in a mouse model of myocardial infarction (Xuan et al., 2001). Subsequent studies have revealed that STAT3 is also involved in the induction of preconditioning by sublethal ischemia in both in vitro and in vivo ischemic neuronal injury models (Kim et al., 2004; Kim et al., 2008; Yagi et al., 2011). In addition, NSCs exposed to interleukin-6, a representative upstream cytokine of the JAK2/STAT3 pathway, showed increased STAT3 activation and improved efficacy of NSC transplantation therapy in a mouse stroke model (Sakata et al., 2012). These findings suggest that STAT3 may also play an important role in HP of NSCs, but this has not been investigated.
The purpose of the present study is to investigate the effect of HP on NSCs against oxidative stress and to test the hypothesis that STAT3 and its related signals play pivotal roles in the HP of NSCs. Using NSCs derived from neonatal mice, we investigated the protective effects of 24-hour HP against oxidative injury due to 12-hour exposure to H2O2. We examined the expression of phospho-STAT3 (Tyr705) after HP to check the activation state of STAT3. In addition, we studied the effects of a STAT3 inhibitor on the neuroprotective effect and the expression of representative STAT3-related signals such as JAK2, VEGF, and cyclin D1.
Methods
The study was not pre-registered, and no randomization was performed to allocate subjects in the study. Neither randomization nor sample size calculation was performed. Inclusion/exclusion criteria are not used in this study.
Animals
C57BL/6 mice (CLEA Japan, Japan) were used for this study, and all animals were treated in accordance with ARRIVE and the University of Yamanashi guidelines. A total of 150 neonatal mice of both sexes from 30 pregnant mice were used in this study. The animal protocols were approved by the University of Yamanashi’s Administrative Panel on Laboratory Animal Care (Approval No. A1-19).
Isolation and Culture of NSCs
NSCs were isolated from the subventricular zones of postnatal day 1 mice as described previously (Wakai et al., 2016). In brief, bilateral subventricular zones were dissected and mechanically dissociated. The cells were collected and suspended in Neurobasal-A medium (Thermo Fisher Scientific, Waltham, MA, USA, #10888022) containing 2% B-27 supplement (Thermo Fisher scientific, #12587010), 2% L-glutamine (Thermo Fisher scientific, # 25030081), 20 ng/mL mouse fibroblast growth factor basic (PeproTech, Rocky Hill, NJ, USA, #45033), and 20 ng/mL mouse epidermal growth factor (PeproTech, #31509). The cells (1.0 × 105 cells/mL) were cultured in standard cell culture conditions in a humidified 5% CO2-95% air incubator at 37°C and grown as neurospheres (western blot analysis and cell viability assay) or adherent monolayers (fluorescence immunostaining and enzyme-linked immunosorbent assay (ELISA)). Both growth factors were added every two days, and cells were passaged weekly. NSCs that had been passaged three to ten times were used for the experiments. To observe the differentiation of NSCs, cells were incubated in medium without growth factors containing 1% bovine serum albumin (BSA) (Sigma-Aldrich, St Louis, MO, USA, #A9576) and were allowed to differentiate for seven days. For the histological study, NSCs were cultured on 8-well chamber slides (Corning, Corning, NY, USA, #354688).
HP
For the induction of HP, NSCs were cultured in standard cell culture conditions in a humidified 5% CO2-95% air incubator at 37°C for 24 hours and then incubated at 37°C in hypoxic conditions (5% O2, 5% CO2, and 90% N2) for 24 hours in a gas-tight humidified chamber (Hypoxia incubator chamber, STEMCELL Technologies, Vancouver, BC, Canada, #27310) as described previously (Wakai et al., 2016). Subsequently, the NSCs were returned to standard conditions for 12 hours.
Oxidative Stress
As lethal oxidative stress, cultured NSCs were exposed to H2O2 for 12 hours. The timing of exposure to H2O2 was 12 hours after HP, and the concentration of H2O2 was 100 μM, 500 μM, and 1 mM. For the STAT3 inhibition experiment, 100 μM H2O2 was used.
Immunocytochemistry
NSCs were fixed in 4% formaldehyde for 15 minutes at 4°C, washed with PBS, and subsequently blocked with 5% bovine serum albumin (BSA) for one hour at room temperature. Following blocking, the samples were incubated with primary antibodies overnight at 4°C. The primary antibodies were anti-mitogen-activated protein 2 (MAP2) (1:1000; Sigma-Aldrich, #M4403), anti-glial fibrillary acidic protein (GFAP) (1:1000; Thermo Fisher Scientific, #130300), anti-neuron-glial antigen 2 (NG2) (1:1000; Abcam, Cambridge, UK, #ab129051), and anti-nestin (1:100; BD Biosciences, Franklin Lakes, NJ, USA, #556309). Then, the samples were incubated with Alexa Fluor 488 antibody (1:2000; Invitrogen, #R37118) for one hour at room temperature. The slides were covered with VECTASHIELD mounting medium with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA, #H-1500) and examined with a fluorescent microscope (Axio Imager, Carl Zeiss, Oberkochen, Germany).
Cell Viability Assay
Cell viability was assessed with a cell proliferation reagent using a water-soluble tetrazolium 1 (WST-1) assay kit (Cell Proliferation Reagent WST-1, Sigma-Aldrich, #5015944001). NSCs were cultured in 96-well plates (Corning, #354657) as neurospheres. To confirm the characteristics of the cultured cells, cell viability was assessed on days 1, 2, 3, 4, and 5. To investigate whether hypoxic conditions enhance cell proliferation, viability was assessed 0, 24, and 48 hours after hypoxia. To examine the effects of oxidative stress with or without HP, the WST-1 assay was performed just after 12-hour exposure to H2O2. Aliquots of media and WST-1 reagent were mixed in a 96-well plate, and absorbance at 450 nm was recorded using a microplate spectrophotometer system (SpectraMax® ABS plus, Molecular Devices, Sunnyvale, CA, USA). Results were analyzed with the Softmax Pro 7 software and are presented as ratios to control values.
Assessment of Cell Injury
Cell injury due to hypoxic conditions and/or oxidative stress was assessed with terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) staining. The timing of the assessment of cell injury was the same as the cell viability assay. TUNEL staining was performed with a commercial kit (In Situ Cell Death Detection Kit Fluorescein, Sigma-Aldrich, #11684795910), according to the manufacturer’s protocol. The slides were covered with VECTASHIELD mounting medium with DAPI. TUNEL- and DAPI-positive cells were counted in four randomly selected high-power fields (×400 magnification) by two counters who were blinded to the experimental conditions. The ratio of TUNEL-positive cells to the number of DAPI-stained nuclei was calculated.
Detection of VEGF
To detect VEGF as a paracrine factor, NSCs were cultured as adherent cells in a 6-cm dish, and the culture medium was collected for analysis 24 hours after HP. VEGF ELISA kits (R&D Systems, Minneapolis, MN, USA, #MMV00) were used, and absorbance at 450 nm was recorded using a microplate spectrophotometer system. The concentration of VEGF in each sample was measured by comparing the results with the standard reagent.
Western Blotting
The changes in the expression of STAT3 and its related signals (JAK2 and cyclin D1) in NSCs were analyzed using western blotting. NSCs were cultured as neurospheres in a 10-cm dish, and the protein samples were collected 24 hours after termination of HP. When investigating the time course of phospho-STAT3 (pSTAT3) and STAT3 expression after HP, the samples were collected 0, 6, 12, 24, and 48 hours after HP. NSCs were treated with cell lysate buffer (Cell Signaling Technology, Beverly, MA, USA, #9803S) and used as whole-cell lysate samples. The protein content of NSCs was calculated using the Pierce Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific, #A53226). Twenty micrograms of the sample were loaded per lane and analyzed with sodium dodecyl sulfate-polyacrylamide-gel electrophoresis on a 10% NuPAGE Bis-Tris gel (Invitrogen, #NP0301BOX) at 200 V, 150 mA, 200 W for 45 minutes, and then immunoblotted to PVDF membranes (Thermo Fisher Scientific, # IB401001). The membranes were blocked for 30 minutes at room temperature with 5% BSA, followed by overnight incubation with the primary antibody at 4℃. The primary antibodies were mouse monoclonal anti-STAT3 (1:1000; Cell Signaling Technology, #9139), rabbit monoclonal anti-pSTAT3 (1:1000; Tyr705) (Cell Signaling Technology, #9145), rabbit monoclonal anti-JAK2 (1:1000; Abcam, #ab108596), rabbit monoclonal anti-phospho-JAK2 (pJAK2) (1:1000; Y1007 + Y1008) (Abcam, #ab32101), rabbit monoclonal anti-cyclin D1 (1:1000; Abcam, #ab16663), and mouse monoclonal anti-β-actin (1:5000; Cell Signaling Technology, #A2228). After incubation with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Cell Signaling Technology, #7076) or anti-rabbit immunoglobulin G (Cell Signaling Technology, #7074), the antigen was detected with Super Signal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific, #34580). Images were captured with a lumino image analyzer (ImageQuantTM LAS 4000, Cytiva, Marlborough, MA, USA), and the results were quantified using Image J.
Inhibition of STAT3
STAT3 was blocked with a STAT3 inhibitor peptide (Sigma-Aldrich, #573096), which competitively inhibits STAT3 dimerization and disrupts STAT3 dimers in vitro by direct interaction with STAT3 monomers (Catalano et al., 2005). NSCs were incubated with 1 μM STAT3 inhibitor peptide administered at the same time as HP initiation.
Statistical Analysis
All data are shown as the mean ± standard error of the mean. Comparisons among multiple groups were analyzed with Tukey’s test. For two-group comparisons, a Student’s t-test was performed. Significance was accepted at p < 0.05. Microsoft Excel 2018 [Version: 16.16.27 (201012)] was used for all statistical analysis. All the experimental results were reanalyzed by a second individual blinded to groups to avoid bias.
Results
Confirmation of Neural Stemness of the Cultured Cells
In the in vitro culture, the cells proliferated and formed spherical free-floating aggregates known as neurospheres over
time (Fig. 1A). Proliferation of the cells was examined with the WST-1 assay, which demonstrated that the cells significantly proliferated over time from day 1 to 5 (n = 16, p < 0.01) and cell viability at day 5 increased three times compared with day 1 (Fig. 1B).
To confirm the multipotency of the cells, they were cultured in differentiation medium containing 1% BSA for seven days and were then immunostained for lineage markers (neuronal progenitor cells; MAP2, mature neurons; GFAP, astrocytes; NG2, oligodendrocytes; nestin, immature cells) (Fig. 1C). Fluorescent staining revealed that some of the immature cells (14.9 ± 10.3%, n = 5) were still stained with nestin and that the cultured cells differentiated into mature neurons (28.3 ± 9.0%, n = 5), astrocytes (47.7 ± 19.4%, n = 5), and oligodendrocytes (2.8 ± 6.2%, n = 5) (Fig. 1D). These data showed that the cultured cells used in the present study were primarily NSCs exhibiting well-preserved proliferation and differentiation potential.
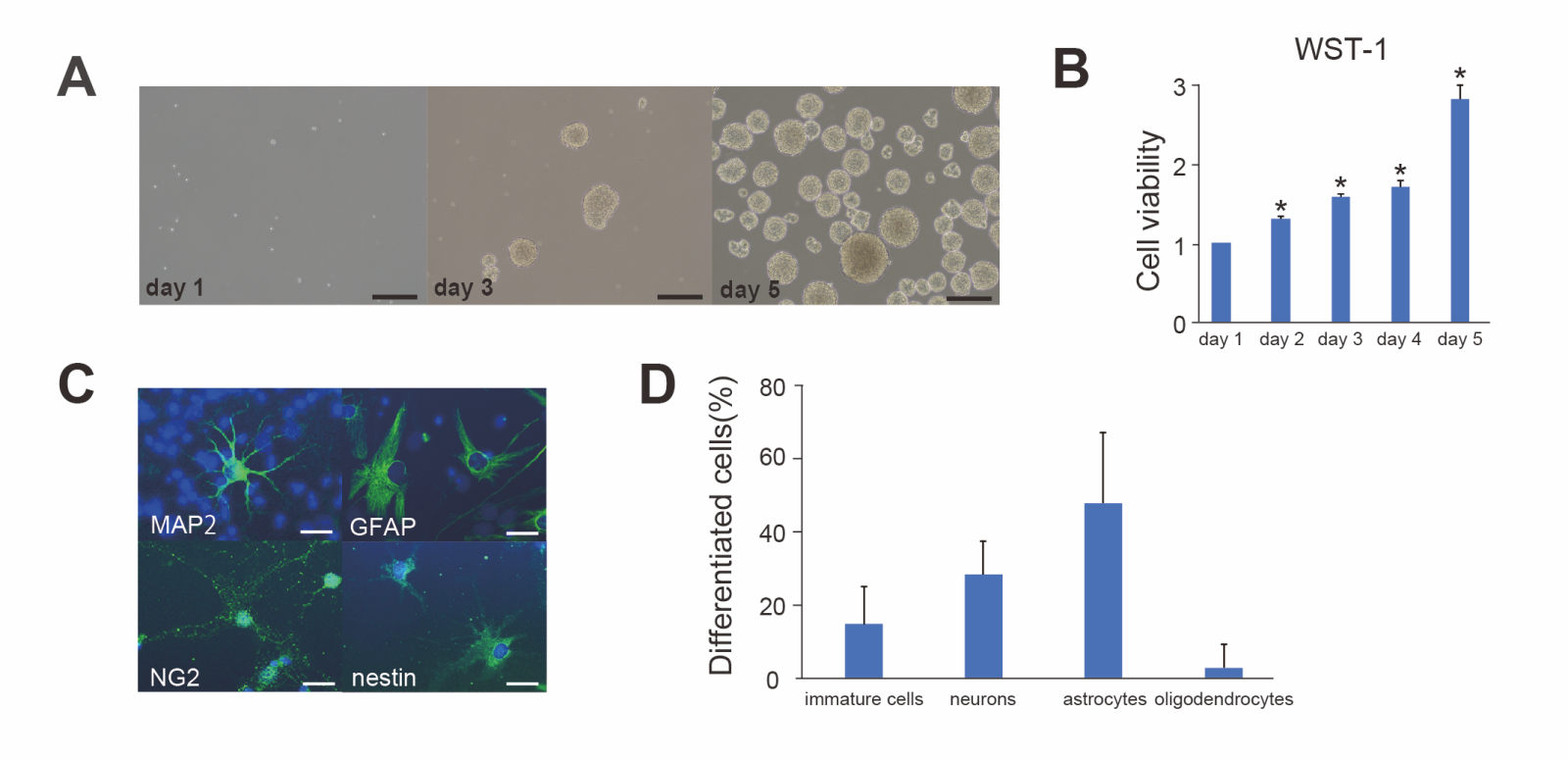
In a new window | Download PPT
Figure 1. Proliferation and differentiation of NSCs (A) Cultured cells proliferated and formed clonal spheres referred to as neurospheres. Scale bar = 100 μm. (B) The water-soluble tetrazolium 1 (WST-1) assay revealed that the cell viability of cultured cells gradually increased from day 1 to 5 (n = 16, *p < 0.01). (C) Differentiated cells were stained with the neuronal marker mitogen-activated protein 2 (MAP2) (green), the astrocytic marker glial fibrillary acidic protein (GFAP) (green), the oligodendrocytic marker neuron-glial antigen 2 (NG2) (green), the immature neural stem cell marker nestin (green), and 4’,6-diamidino-2-phenylindole (DAPI) (blue). The cells diferentiated into neurons, astrocytes, and oligodendrocytes, and a portion of the cells remained immature. Scale bar = 10 μm. (D) The rates of differentiated cells (n = 5).
NSC Injury Induced by Oxidative Stress with Hydrogen Peroxide
To investigate the effects of oxidative stress, NSCs were treated with H2O2 (100 μM, 500 μM, and 1 mM) as lethal oxidative stress, and TUNEL and WST-1 assays were performed 12 hours after the stress. H2O2 significantly increased NSC injury, as indicated by TUNEL staining, in a dose-dependent manner (n = 6, p < 0.05 or 0.01) (Fig. 2B and C). Conversely, the WST-1 assay revealed that the cell viability of NSCs was significantly reduced by exposure to H2O2 in a dose-dependent manner compared to control (n = 6, p < 0.05 or 0.01) (Fig. 2D).
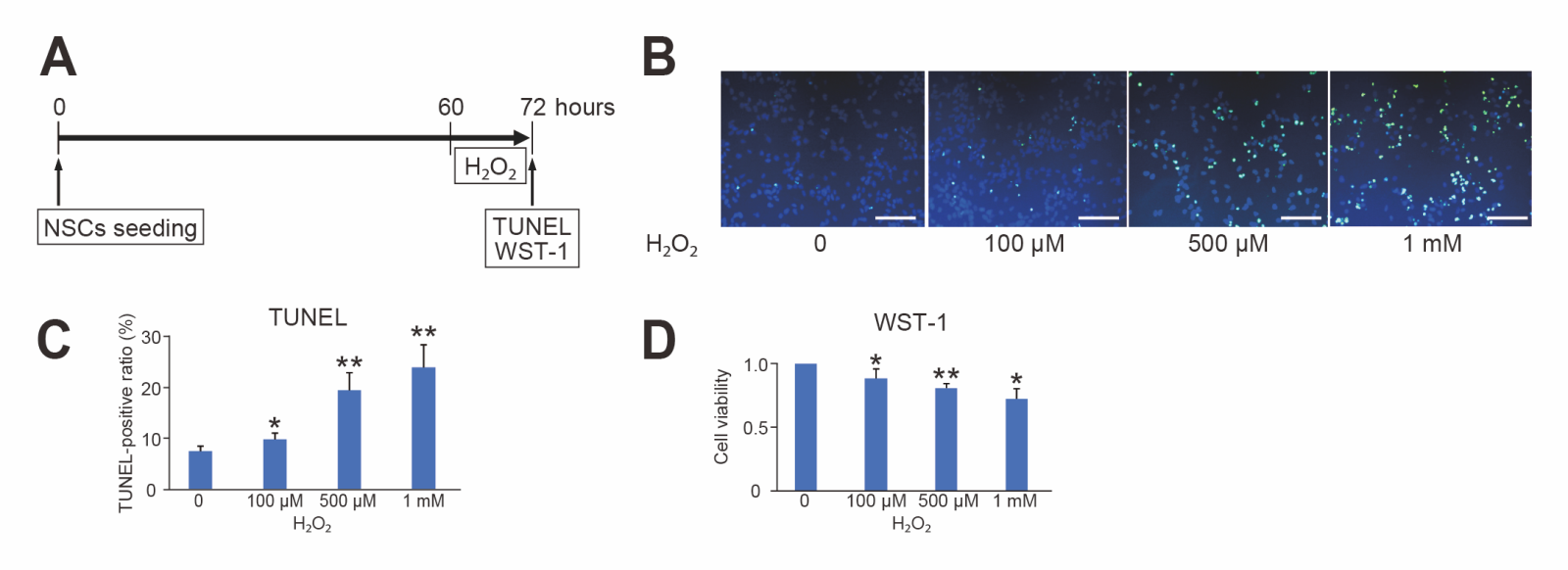
In a new window | Download PPT
Figure 2. Oxidative injury induced by hydrogen peroxide. (A) Timeline for H2O2 and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (B) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). Scale bar = 100 μm. (C) Cell counting showed that H2O2 increased the TUNEL-positive ratio of NSCs in a dose-dependent manner (n = 6, *p < 0.05, **p < 0.01). (D) The WST-1 assay revealed that H2O2 reduced the cell viability of NSCs in a dose-dependent manner (n = 6, *p < 0.05, **p < 0.01).
Effects of 24-Hour Hypoxia on NSCs
The effects of hypoxic conditions on cultured NSCs were examined. TUNEL-positive cells were rarely observed 0, 24, and 48 hours after 24-hour hypoxia (Fig. 3B). On the other hand, the cell viability of NSCs as measured by the WST-1 assay significantly increased after hypoxia (n = 6, p < 0.05 or 0.01) (Fig. 3C).
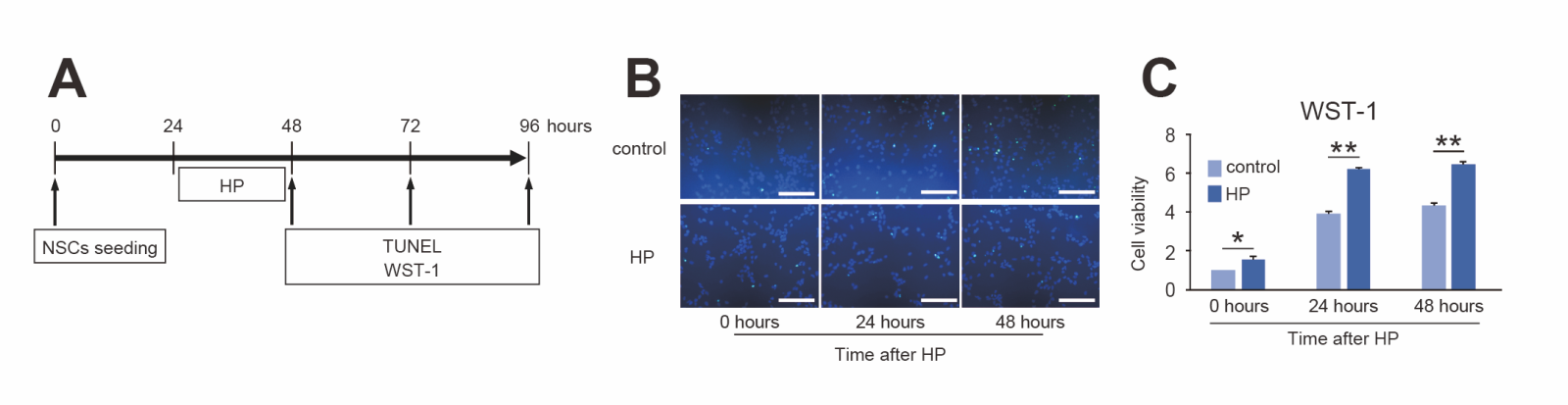
In a new window | Download PPT
Figure 3. Effects of hypoxic preconditioning (HP) on NSCs. (A) Timeline for HP and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (B) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). TUNEL-positive cells were rarely observed 0, 24, and 48 hours after HP. Scale bar = 100 μm. (C) The WST-1 assay showed that HP significantly increased cell viability 0 to 48 h after HP compared with control (n = 6, *p < 0.05, **p < 0.01).
We then investigated whether 24-hour HP enhanced the resilience of NSCs to lethal oxidative stress. The TUNEL assay revealed that HP significantly decreased cell death 12 hours after H2O2 exposure (n = 6, p < 0.05 or 0.01) (Fig. 4B and C). Further, the WST-1 assay showed that HP significantly prevented the decrease in cell viability due to H2O2 exposure (n = 5, p < 0.05) (Fig. 4D).
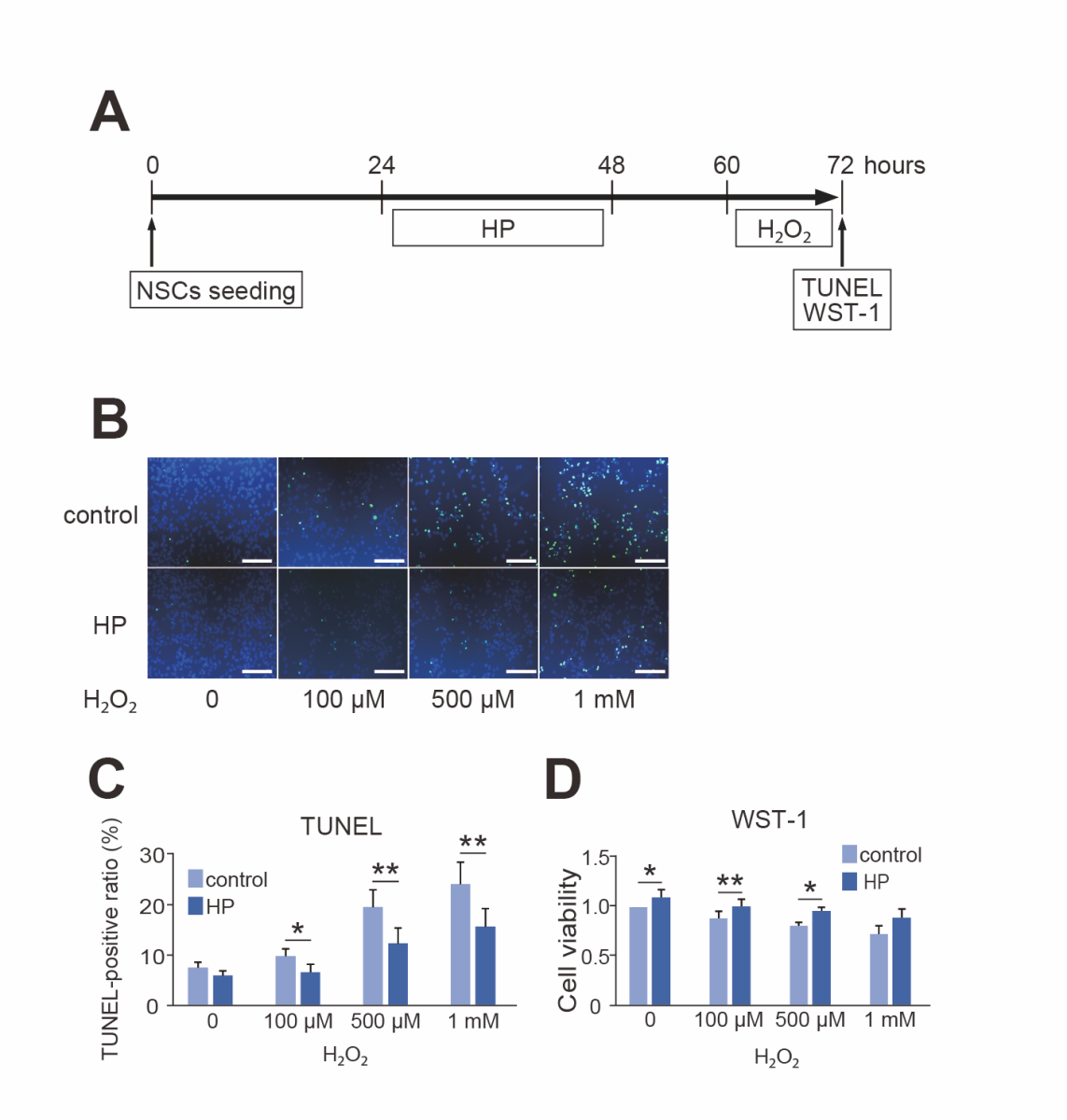
In a new window | Download PPT
Figure 4. Protective effect of hypoxic preconditioning (HP) against oxidative stress. (A) Timeline for HP, H2O2, and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (B) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). HP reduced the number of TUNEL-positive cells after exposure to H2O2. Scale bar = 100 μm. (C) Cell counting showed that HP decreased the TUNEL-positive ratio of NSCs after exposure to H2O2 (n = 6, *p < 0.05, **p < 0.01). (D) The WST-1 assay revealed that HP suppressed the decrease in cell viability due to H2O2 compared with control (n = 5, *p < 0.05, **p < 0.01).
HP did not change the differentiation ability of NSCs (mature neurons, 27.7 ± 1.5%; astrocytes, 45.0 ± 25.4%; oligodendrocytes, 3.5 ± 0.2%) compared with the controls.
Phospho-STAT3 Expression in NSCs after HP and the Effect of the STAT3 Inhibitor
To reveal the molecular mechanism of HP, pSTAT3 expression in NSCs after HP was investigated. Western blot analysis revealed that the expression of pSTAT3 (Tyr705) was significantly increased 0 to 48 hours after HP compared to control (n = 6, p < 0.05 or 0.01). In contrast, the total STAT3 expression was not changed (n = 3) (Fig. 5B). The STAT3 inhibitor peptide significantly decreased pSTAT3 (Tyr705) expression after HP (n = 7, p < 0.01) (Fig. 5D). It reversed the protective effects of HP on NSC injury due to lethal oxidative stress (100 μM H2O2) (Fig. 5F and G). In addition, inhibition of pSTAT3 decreased the cell viability of preconditioned NSCs in the WST-1 assay (Fig. 5H).
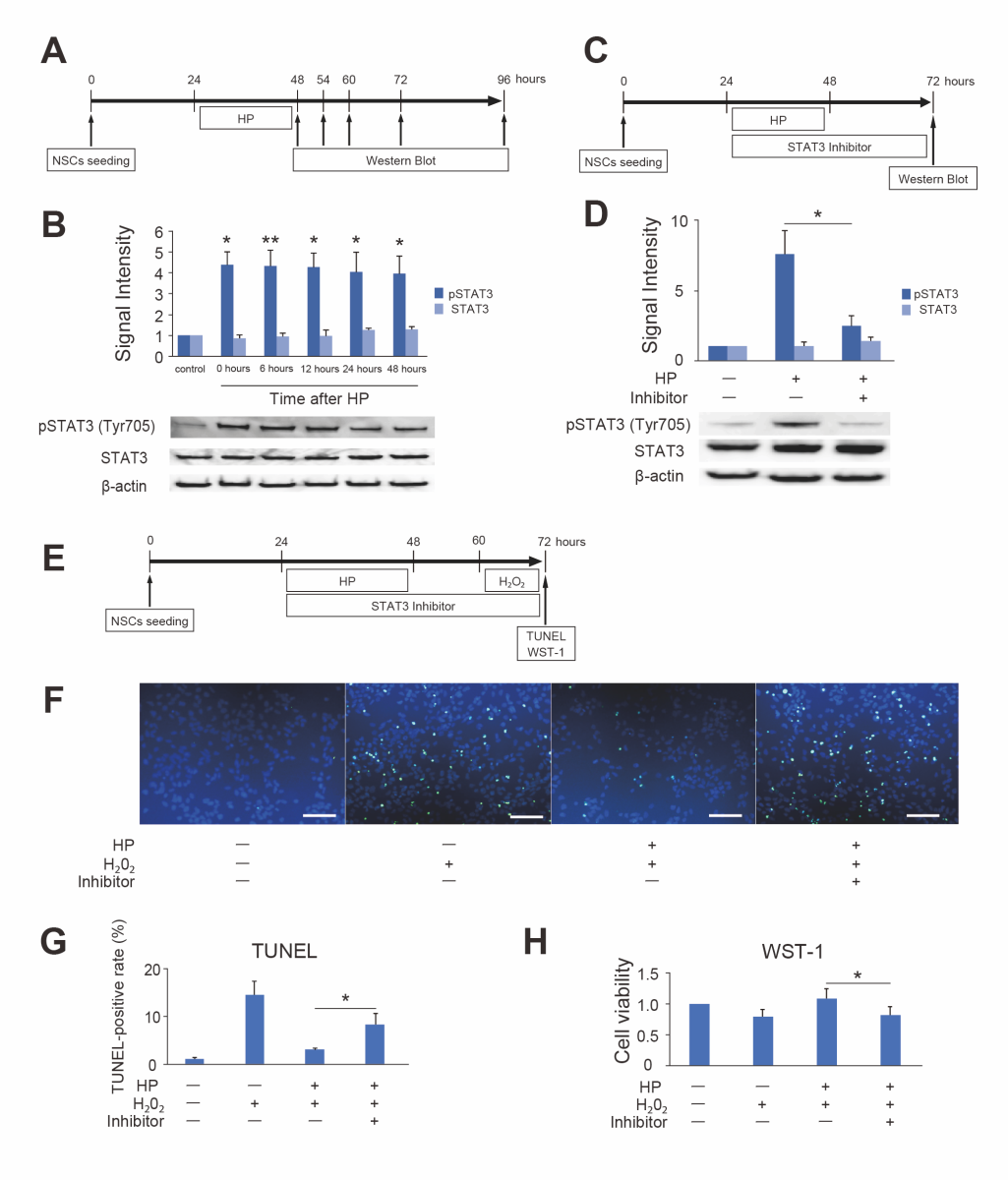
In a new window | Download PPT
Figure 5. Change in expression of phosphorylated STAT3 (pSTAT3) (Tyr705) after hypoxic preconditioning (HP) and effects of a STAT3 inhibitor (A) Timeline for HP and the timing of Western blot analysis. (B) Western blot analysis revealed that pSTAT3 (Tyr705) expression in NSCs significantly increased 0 to 48 hours after HP (n = 6, *p < 0.05, **p < 0.01). Total STAT3 expression was not changed (n = 3). β-actin was used as an internal control. (C) Timeline for HP, the STAT3 inhibitor, and the timing of Western blot analysis. (D) The STAT3 inhibitor peptide suppressed the upregulation of pSTAT3 (Tyr705) in NSCs after HP (n = 7, *p < 0.05). Total STAT3 expression was not changed. β-actin was used as an internal control. (E) Timeline for HP, the STAT3 inhibitor, H2O2, and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (F) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). The STAT3 inhibitor peptide increased the number of TUNEL-positive cells after exposure to H2O2, even with HP. Scale bar = 100 μm. (G) Cell counting showed that the STAT3 inhibitor peptide increased the TUNEL-positive ratio after exposure to H2O2 even with HP (n = 5, *p < 0.05). (H) The WST-1 assay showed that the STAT3 inhibitor peptide decreased the cell viability of preconditioned NSCs after exposure to H2O2 (n = 5, *p < 0.05).
The differentiation ability of NSCs (mature neurons, 28.4 ± 8.3%; astrocytes, 38.0 ± 14.7%; oligodendrocytes, 1.6 ± 0.3%) was unaffected by the STAT3 inhibitor peptide.
Effects of STAT3 Inhibitors on STAT3-Related Signals after HP
The STAT3-related signaling factors in NSCs after HP were elucidated using western blot analysis (Fig. 6B). The expression levels of JAK2 and pJAK2, a well-known upstream factor of the STAT3 pathway, were significantly elevated 24 hours after HP, and the STAT3 inhibitor further increased the total expression level of JAK2 (n = 7, p < 0.05). However, the expression level of pJAK2 was not elevated by the STAT3 inhibitor (Fig. 6C).
Regarding molecules downstream of STAT3 signals, cyclin D1 expression was increased after HP and was significantly inhibited by the STAT3 inhibitor (n = 7, p < 0.05 or 0.01) (Fig. 6D). Moreover, HP increased the VEGF concentration in the culture medium as seen with ELISA, and the STAT3 inhibitor suppressed this increase (n = 7, p < 0.05) (Fig. 6E).
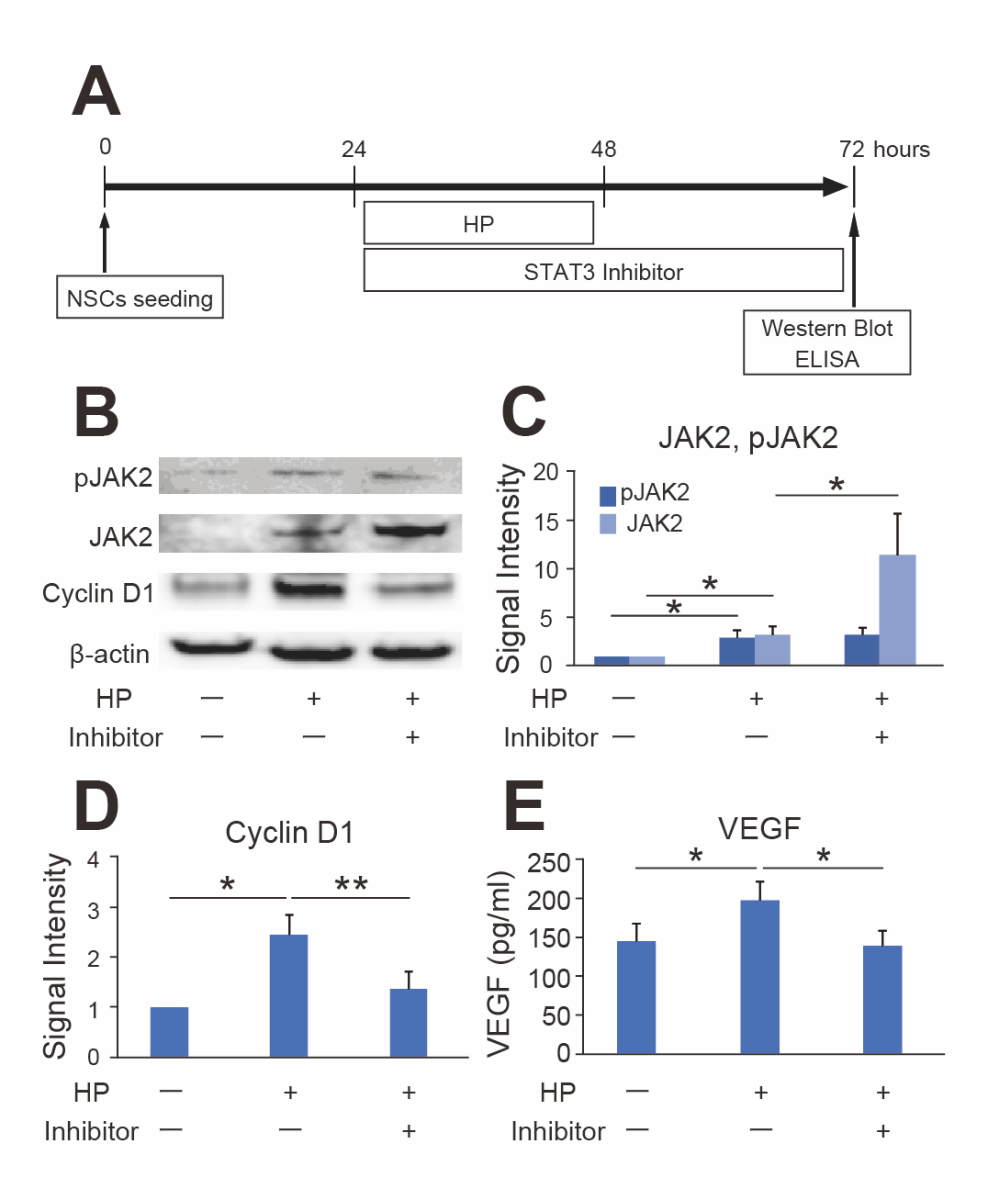
In a new window | Download PPT
Figure 6. Expression changes in STAT3-related signals. (A) Timeline for HP, STAT3 inhibitor, and the timing of Western blot and Enzyme-linked immunosorbent assay (ELISA). (B-D) Western blot analysis showed that the expression levels of phosphorylated JAK2 (pJAK2), JAK2, and cyclin D1 significantly increased 24 hours after HP. The STAT3 inhibitor peptide significantly increased JAK2 expression and decreased cyclin D1 expression after HP (n = 7, *p < 0.05, **p < 0.01). β-actin was used as an internal control. (E) The ELISA showed that HP significantly increased VEGF. The STAT3 inhibitor peptide suppressed the upregulation of VEGF after HP (n = 7, *p < 0.05).
Discussion
Preconditioning is a phenomenon in which tolerance to later lethal stimuli is acquired after exposure to non-lethal stimuli (Murry et al., 1986; Kitagawa et al., 1990; Hirayama et al., 2015). Several reports have shown that HP can markedly improve the tolerance of treated subjects to more severe insults (Liu et al., 2000; Theus et al., 2008) and has received attention in recent years as a strategy for enhancing the therapeutic effect of NSC transplantation after stroke. However, the mechanism of HP on NSCs is not well understood. The major findings of the present study are: (1) 24-hour HP enhanced the cell viability of NSCs without any harmful effects; (2) HP protected NSCs against oxidative stress induced by exposure to H2O2; (3) HP activated JAK2 and STAT3 throughout preconditioning, enhancing the production of VEGF and the expression of cyclin D1; (4) the STAT3 inhibitor suppressed the upregulation of VEGF and cyclin D1 and reversed the protective effect of HP.
STAT3 is a transcription factor with neuroprotective effects that play an important role in preconditioning settings such as ischemia, hypoxia, oxygen-glucose deprivation, and chemical substances (Darnell, 1997; Bromberg and Darnell, 2000; Xuan et al., 2001; Levy and Lee, 2002; Kim et al., 2004; Kim et al., 2008; Wang et al., 2010; Yagi et al., 2011; Sakata et al., 2012; Cheng et al., 2014; Zhao et al., 2019). In the present study, STAT3 phosphorylation (Tyr705) was enhanced for 48 hours after HP, indicating that HP activated STAT3 during the tolerance period. In addition, the tolerance effect of HP was attenuated by the STAT3 inhibitor peptide that contains the STAT3 SH2 domain binding peptide, PYLKTK. This peptide competitively inhibits STAT3 dimerization, disrupts STAT3 dimers in vitro by directly interacting with STAT3 monomers, and suppresses the expression of pSTAT3 (Tyr705) (Catalano et al., 2005). Western blot analysis in the present study showed that the inhibitor peptide attenuated the expression of pSTAT3, revealing that the dose of the inhibitor used in this study was adequate to suppress STAT3 activation effectively. According to these results, the present study clearly showed for the first time that STAT3 plays a crucial role in the HP of NSCs.
JAK2, the major upstream signaling factor of the JAK2/STAT3 pathway, is a crucial factor involved in signaling through various cytokine receptors and phosphorylates STAT3 upon activation (Hou et al., 2018). In this study, we confirmed that HP increased the expression and phosphorylation of JAK2 in accordance with enhanced phosphorylation of STAT3. Moreover, inhibition of STAT3 increased the expression of JAK2, which may be promoted by inhibiting negative feedback (Yu et al., 2017). Therefore, the data suggest that JAK2 may be a significant factor in activating STAT3 in HP of NSCs.
Because activation of STAT3 promotes transcription of genes mainly involved in proliferation, survival, and differentiation, we examined VEGF and cyclin D1 as representative STAT3 downstream factors. We demonstrated that these factors were upregulated by HP and suppressed by the STAT3 inhibitor, suggesting that they act downstream of STAT3 in the HP-induced protection of NSCs. VEGF is a well-known angiogenic factor that mainly binds to its receptor on the surface of vascular endothelial cells as a ligand that stimulates cell division, migration, and differentiation and enhances vascular permeability of microvessels. In neuronal cells, VEGF confers neuroprotection in addition to angiogenesis (Geiseler and Morland, 2018). VEGF released from retinal ganglion cells promotes their survival, indicating autocrine VEGF neuroprotection of retinal ganglion cells. In parallel, VEGF produced by mixed retinal cells or mesenchymal stem cells exerts paracrine neuroprotection on retinal ganglion cells (Froger et al., 2020). An autocrine mechanism was also suggested to explain the neuroprotective function of VEGF on embryonic cortical neurons (Ogunshola et al., 2002). Taken together, these observations suggest that the neuroprotective function of upregulated VEGF from NSCs contributed to the protective effect of HP via an autocrine/paracrine mechanism in the present study.
Ischemic preconditioning of liver cells promotes the expression of cyclin D1 and leads to their proliferation during early ischemic reperfusion, suggesting the possibility of a protective mechanism against ischemia-reperfusion injury (Cai et al., 2006). Another study reported that garcinone D, a natural xanthone isolated from mangosteen, increases cyclin D1 expression via tyrosine phosphorylation of STAT3, which promotes the proliferation of NSCs and enhances the number of cells in the S phase (Yang et al., 2016). In the present study, the WST-1 assay for proliferation showed that HP had a cell proliferative effect on NSCs. Because of the cell proliferation efficacy of cyclin D1 mentioned above, upregulation of cyclin D1 via STAT3 activation may enhance cell activity and contribute to the acquisition of HP in NSCs.
The present study has some limitations. Oxidative stress was assumed to be the most harmful factor of the host environment after stroke in this experiment, but other factors such as excitotoxicity, calcium overload, mitochondrial dysfunction, and inflammation are also present after stroke. In addition to oxidative stress, the effect of HP and the role of STAT3 on other such stresses should be further examined. In addition, because our final purpose is to improve the efficacy of cell transplantation therapy, the roles of HP and STAT3 in an in vivo model should be investigated in a future study. Finally, experiments using inhibitors of VEGF and cyclin D1 proteins should be performed because the direct effects of these proteins on HP have not been demonstrated, and examinations of other downstream signals of STAT3 should be subject to further studies.
In conclusion, HP induces tolerance to oxidative stress by activating STAT3 in NSCs. The protective function of autocrine/paracrine VEGF and cell proliferation effects of cyclin D1 likely contribute to the acquisition of preconditioning as downstream mediators of the JAK2/STAT3 pathway (Fig.7). Activation of this pathway may be a treatment strategy to improve the efficacy of cell therapy in patients with stroke.
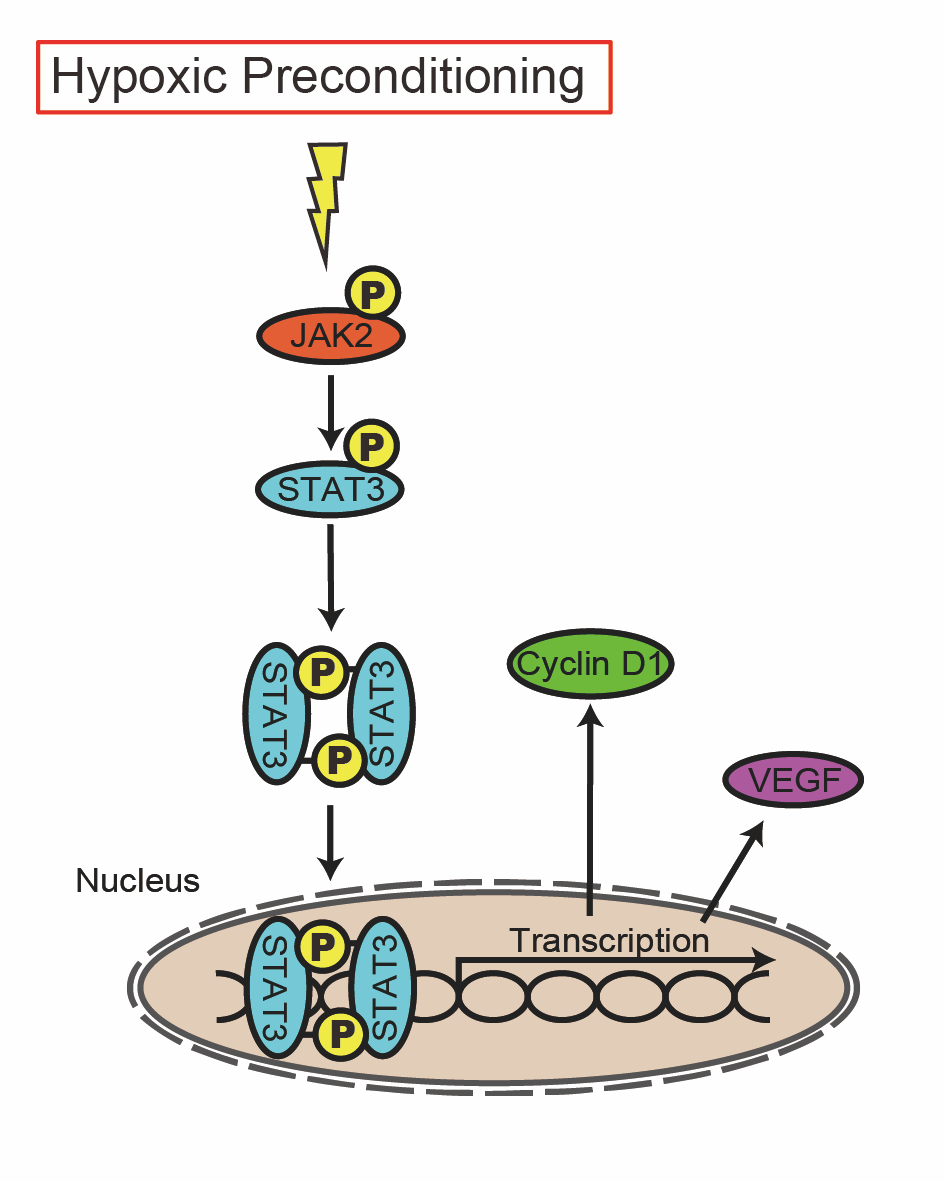
In a new window | Download PPT
Figure 7. Graphical Abstract. The role of the JAK2/STAT3 pathway in NSCs. HP induces tolerance to oxidative stress through activation of JAK2/STAT3 in NSCs. The protective function of autocrine/paracrine VEGF and cell proliferation effects of cyclin D1 likely contribute to the acquisition of preconditioning as downstream mediators of the pathway.
Conflict of interest disclosure
The authors declare that they have no conflicts of interest. We hereby disclose that Hiroyuki Kinouchi, listed as an author of this research paper, also holds a position as a member of the editorial board of Conditioning Medicine. He has not participated in any aspect of the peer review or editorial decision-making process for this paper. All review processes have been independently conducted by third parties.
Acknowledgments
This work was supported financially by the Japan Society for the Promotion of Science KAKENHI (Grant Numbers JP 16K10715 and 19H03769).
References
Norito Fukuda1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Hideyuki Yoshioka1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Takuma Wakai1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Toru Tateoka1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Koji Hashimoto1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Kazuya Kanemaru1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Hiroyuki Kinouchi1
1Department of Neurosurgery, Graduate School of Medicine, Faculty of Medicine, University of Yamanashi, Yamanashi, Japan.
Corresponding author:
Hideyuki Yoshioka
Email: hidey@yamanashi.ac.jp
In a new window | Download PPT
Figure 1. Proliferation and differentiation of NSCs (A) Cultured cells proliferated and formed clonal spheres referred to as neurospheres. Scale bar = 100 μm. (B) The water-soluble tetrazolium 1 (WST-1) assay revealed that the cell viability of cultured cells gradually increased from day 1 to 5 (n = 16, *p < 0.01). (C) Differentiated cells were stained with the neuronal marker mitogen-activated protein 2 (MAP2) (green), the astrocytic marker glial fibrillary acidic protein (GFAP) (green), the oligodendrocytic marker neuron-glial antigen 2 (NG2) (green), the immature neural stem cell marker nestin (green), and 4’,6-diamidino-2-phenylindole (DAPI) (blue). The cells diferentiated into neurons, astrocytes, and oligodendrocytes, and a portion of the cells remained immature. Scale bar = 10 μm. (D) The rates of differentiated cells (n = 5).
In a new window | Download PPT
Figure 2. Oxidative injury induced by hydrogen peroxide. (A) Timeline for H2O2 and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (B) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). Scale bar = 100 μm. (C) Cell counting showed that H2O2 increased the TUNEL-positive ratio of NSCs in a dose-dependent manner (n = 6, *p < 0.05, **p < 0.01). (D) The WST-1 assay revealed that H2O2 reduced the cell viability of NSCs in a dose-dependent manner (n = 6, *p < 0.05, **p < 0.01).
In a new window | Download PPT
Figure 3. Effects of hypoxic preconditioning (HP) on NSCs. (A) Timeline for HP and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (B) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). TUNEL-positive cells were rarely observed 0, 24, and 48 hours after HP. Scale bar = 100 μm. (C) The WST-1 assay showed that HP significantly increased cell viability 0 to 48 h after HP compared with control (n = 6, *p < 0.05, **p < 0.01).
In a new window | Download PPT
Figure 4. Protective effect of hypoxic preconditioning (HP) against oxidative stress. (A) Timeline for HP, H2O2, and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (B) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). HP reduced the number of TUNEL-positive cells after exposure to H2O2. Scale bar = 100 μm. (C) Cell counting showed that HP decreased the TUNEL-positive ratio of NSCs after exposure to H2O2 (n = 6, *p < 0.05, **p < 0.01). (D) The WST-1 assay revealed that HP suppressed the decrease in cell viability due to H2O2 compared with control (n = 5, *p < 0.05, **p < 0.01).
In a new window | Download PPT
Figure 5. Change in expression of phosphorylated STAT3 (pSTAT3) (Tyr705) after hypoxic preconditioning (HP) and effects of a STAT3 inhibitor (A) Timeline for HP and the timing of Western blot analysis. (B) Western blot analysis revealed that pSTAT3 (Tyr705) expression in NSCs significantly increased 0 to 48 hours after HP (n = 6, *p < 0.05, **p < 0.01). Total STAT3 expression was not changed (n = 3). β-actin was used as an internal control. (C) Timeline for HP, the STAT3 inhibitor, and the timing of Western blot analysis. (D) The STAT3 inhibitor peptide suppressed the upregulation of pSTAT3 (Tyr705) in NSCs after HP (n = 7, *p < 0.05). Total STAT3 expression was not changed. β-actin was used as an internal control. (E) Timeline for HP, the STAT3 inhibitor, H2O2, and the timing of terminal deoxynucleotidyl transferase-mediated uridine 5’-triphosphate-biotin nick end labeling (TUNEL) and water-soluble tetrazolium 1 (WST-1) assessment. (F) Representative photomicrographs of TUNEL staining. Live cells were stained with DAPI (blue), and injured cells were stained with TUNEL (green). The STAT3 inhibitor peptide increased the number of TUNEL-positive cells after exposure to H2O2, even with HP. Scale bar = 100 μm. (G) Cell counting showed that the STAT3 inhibitor peptide increased the TUNEL-positive ratio after exposure to H2O2 even with HP (n = 5, *p < 0.05). (H) The WST-1 assay showed that the STAT3 inhibitor peptide decreased the cell viability of preconditioned NSCs after exposure to H2O2 (n = 5, *p < 0.05).
In a new window | Download PPT
Figure 6. Expression changes in STAT3-related signals. (A) Timeline for HP, STAT3 inhibitor, and the timing of Western blot and Enzyme-linked immunosorbent assay (ELISA). (B-D) Western blot analysis showed that the expression levels of phosphorylated JAK2 (pJAK2), JAK2, and cyclin D1 significantly increased 24 hours after HP. The STAT3 inhibitor peptide significantly increased JAK2 expression and decreased cyclin D1 expression after HP (n = 7, *p < 0.05, **p < 0.01). β-actin was used as an internal control. (E) The ELISA showed that HP significantly increased VEGF. The STAT3 inhibitor peptide suppressed the upregulation of VEGF after HP (n = 7, *p < 0.05).
In a new window | Download PPT
Figure 7. Graphical Abstract. The role of the JAK2/STAT3 pathway in NSCs. HP induces tolerance to oxidative stress through activation of JAK2/STAT3 in NSCs. The protective function of autocrine/paracrine VEGF and cell proliferation effects of cyclin D1 likely contribute to the acquisition of preconditioning as downstream mediators of the pathway.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 6018 | 23 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA