Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Mechanisms underlying diabetic cardiomyopathy: From pathophysiology to novel therapeutic targets
Time:2020-05-05
Number:11550
Author Affiliations

Conditioning Medicine 2020. 3(2):82-97.
Abstract
Diabetic cardiomyopathy (DC) is defined as a clinical condition of cardiac dysfunction that occurs in the absence of coronary atherosclerosis, valvular disease, and hypertension in patients with diabetes mellitus (DM). Despite the increasing worldwide prevalence of DC, due to the global epidemic of DM, the underlying pathophysiology of DC has not been fully elucidated. In addition, the clinical criteria for diagnosing DC have not been established, and specific therapeutic options are not currently available. The current paradigm suggests the impaired cardiomyocyte function arises due to a number of DM-related metabolic disturbances including hyperglycemia, hyperinsulinemia, and hyperlipidemia, which lead to diastolic dysfunction and signs and symptoms of heart failure. Other factors, which have been implicated in the progression of DC, include mitochondrial dysfunction, increased oxidative stress, impaired calcium handling, inflammation, and cardiomyocyte apoptosis. Herein, we review the current theories surrounding the occurrence and progression of DC, and discuss the recent advances in diagnostic methodologies and therapeutic strategies. Moreover, apart from conventional animal DC models, we highlight alternative disease models for studying DC such as the use of patient-derived human induced pluripotent stem cells (hiPSCs) for studying the mechanisms underlying DC. The ability to obtain hiPSC-derived cardiomyocytes from DM patients with a DC phenotype could help identify novel therapeutic targets for preventing and delaying the progression of DC, and for improving clinical outcomes in DM patients.
Keywords: Diabetic cardiomyopathy; hyperglycemia; metabolism; mitochondria; human induced pluripotent stem cells (hiPSCs); disease modeling
Abstract
Diabetic cardiomyopathy (DC) is defined as a clinical condition of cardiac dysfunction that occurs in the absence of coronary atherosclerosis, valvular disease, and hypertension in patients with diabetes mellitus (DM). Despite the increasing worldwide prevalence of DC, due to the global epidemic of DM, the underlying pathophysiology of DC has not been fully elucidated. In addition, the clinical criteria for diagnosing DC have not been established, and specific therapeutic options are not currently available. The current paradigm suggests the impaired cardiomyocyte function arises due to a number of DM-related metabolic disturbances including hyperglycemia, hyperinsulinemia, and hyperlipidemia, which lead to diastolic dysfunction and signs and symptoms of heart failure. Other factors, which have been implicated in the progression of DC, include mitochondrial dysfunction, increased oxidative stress, impaired calcium handling, inflammation, and cardiomyocyte apoptosis. Herein, we review the current theories surrounding the occurrence and progression of DC, and discuss the recent advances in diagnostic methodologies and therapeutic strategies. Moreover, apart from conventional animal DC models, we highlight alternative disease models for studying DC such as the use of patient-derived human induced pluripotent stem cells (hiPSCs) for studying the mechanisms underlying DC. The ability to obtain hiPSC-derived cardiomyocytes from DM patients with a DC phenotype could help identify novel therapeutic targets for preventing and delaying the progression of DC, and for improving clinical outcomes in DM patients.
Keywords: Diabetic cardiomyopathy; hyperglycemia; metabolism; mitochondria; human induced pluripotent stem cells (hiPSCs); disease modeling
1. Introduction
Heart failure (HF) remains one of the leading causes of death and disability worldwide, a major cause of which is related to diabetes mellitus (DM) (Gustafsson et al., 2004; Maisch et al., 2011; Shah et al., 2015). The prevalence of DM has increased drastically over the past decade, reaching 9.7% among US adults with type 2 diabetes mellitus (T2DM) and accounting for 91% of cases (Xu et al., 2018). Patients with DM are ~ 2 to 5 fold more likely to develop HF (Dei Cas et al., 2015; Jia et al., 2018), which is considered to be the leading clinical manifestation of cardiovascular disease (CVD) in DM patients (Shah et al., 2015). This brought to the fore the recognition of diabetic cardiomyopathy (DC) as a distinct cardiac disease process and cause of HF in DM patients (Rubler et al., 1972).
The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) Guideline for the Management of Heart Failure (Yancy et al., 2013; Yancy et al., 2017) and European Society of Cardiology (ESC) Guidelines on DM, pre-diabetes, and cardiovascular diseases developed in collaboration with the European Association for the Study of Diabetes (EASD) (Cosentino et al., 2019), have defined DC as a clinical condition of ventricular dysfunction that occurs in the absence of coronary atherosclerosis, valvular disease, and hypertension in patients with DM. Researchers have recently classified DC as a four-staged process with early manifestations of diastolic dysfunction and left ventricular hypertrophy (LVH), and subsequent progression to systolic dysfunction and HF (Mizamtsidi et al., 2016; Lorenzo-Almoros et al., 2017). Currently, there are no well-defined clinical criteria for diagnosing DC, nor are there specific disease management strategies, and this can, in part, be attributed to the poor understanding of the mechanisms underlying the disease.
Several animal models have been developed to recapitulate the diabetic condition, and these manifest the known diabetes-related metabolic disturbances of hyperglycemia, hyperinsulinemia, and hyperlipidemia, which are known to mediate the cardiac dysfunction that occurs in DM (Jia et al., 2018). However, due to obvious differences in biology and physiology between animal models and patients, the former may not accurately recapitulate a complex multisystem disease such as DM (Jimenez-Tellez and Greenway, 2019). An alternative approach for studying the pathophysiological mechanisms underlying DC, especially on the cellular level, relies on the use of robust cellular models. Fortunately, the discovery of human induced pluripotent stem cells (hiPSCs), from which cardiomyocytes can be derived in vitro, has emerged as a promising tool to model DC.
In this article, we review the current diagnostic and therapeutic strategies for DC, and highlight the metabolic perturbations and cellular mechanisms that underlie the onset and progression of DC. We also highlight the application of hiPSCs in modeling DC to explore the underlying pathophysiology, which should allow the identification of novel therapeutic targets for preventing and delaying the progression of DC, and improving clinical outcomes in DM patients.
2. Pathophysiology of diabetic cardiomyopathy
Diabetes-related metabolic disturbance
DC is considered a complex entity arising from multiple pathophysiological mechanisms (Figure 1) including hyperglycemia, insulin resistance, microvascular changes, inflammation, and autonomic neuropathy (Maisch et al., 2011; Perrone-Filardi et al., 2015; Dillmann, 2019). The actual etiological pathophysiology, however, is not completely understood. DC has been described as having two phenotypes - dilated/HF with reduced left ventricular ejection fraction (HFrEF) and restrictive/HF with preserved left ventricular ejection fraction (HFpEF), with different pathogenic origins of myocardial dysfunction and mechanisms that contribute to myocardial remodeling (Seferovic and Paulus, 2015). Interestingly, the restrictive profile seems to be the more prevalent form in patients with T2DM, where hyperglycemia and hypertriglyceridemia are found to be contributing factors.
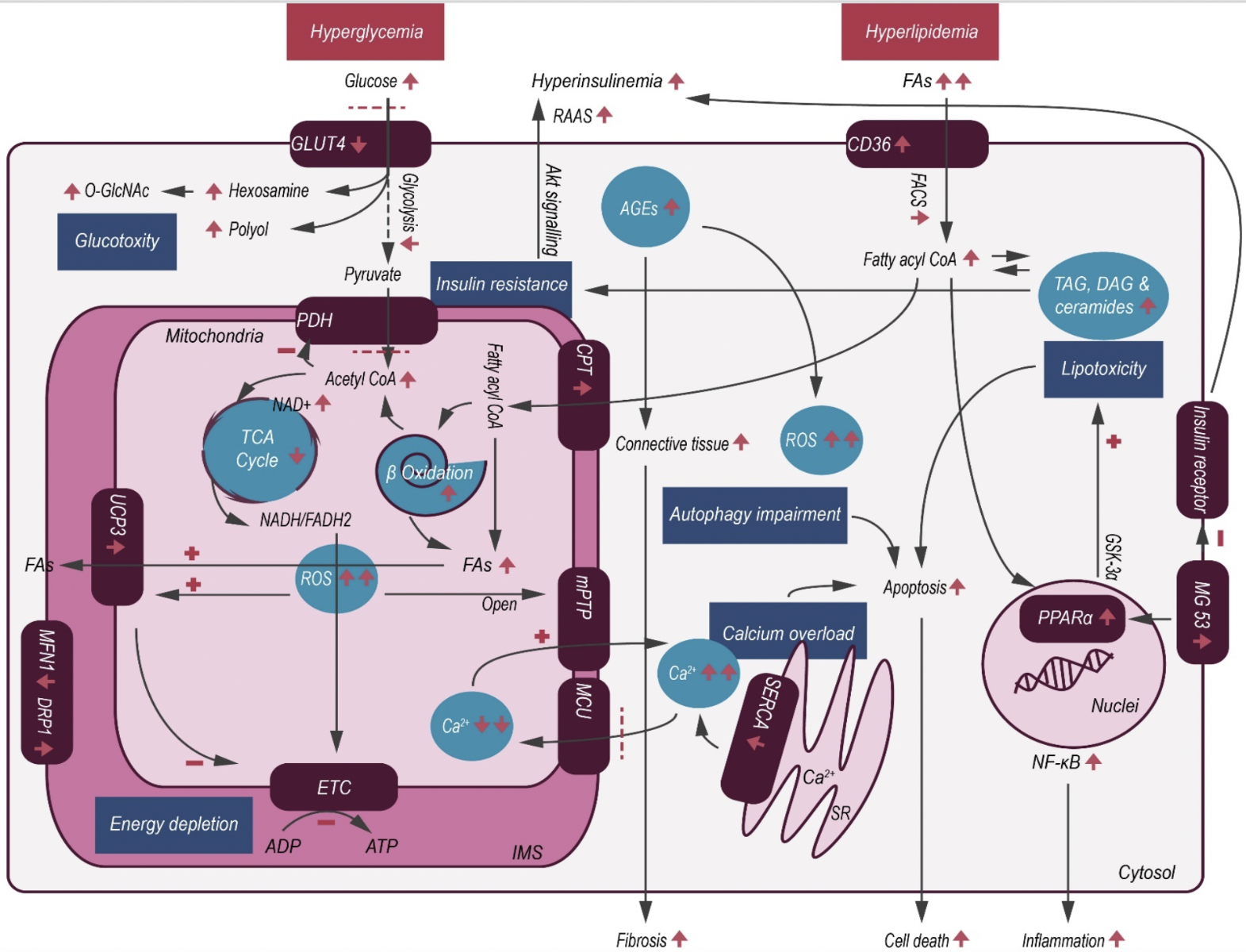
In a new window | Download PPT
Figure 1: Schematic illustration of cardiac metabolic alterations in DC. In T2DM, hyperglycemia and hyperlipidemia cause a series of changes within the myocardium, resulting in decreased glucose uptake but with concomitant increase in FAO. Excessive FA metabolites and non-glycolytic glucose products mediate lipotoxicity and glucotoxity, respectively. As a result of maladaptive metabolism, increased intracellular ROS activates mitochondrial uncoupling, making ATP production less efficient, and also causes opening of the mPTP, thereby initiating apoptosis. Abbreviations: AGE- advanced glycation end product; CPT- carnitine palmitoyltransferase; CoA- coenzyme A; FACS- fatty acyl-coenzyme A synthetase; CD36- fatty acid translocase; DAG- diacylglycerol; ETC- electron Transport Chain; FA- fatty acid; GLUT4- glucose transporters 4; GSK-3α- glycogen synthase kinase-3α; IMS- inner membrane space; MG 53- mitsugumin 53; mPTP- mitochondrial permeability transition pore; MCU- mitochondrial calcium uniporter; NAD+- nicotinamide adenine dinucleotide; NADH/FADH2- nicotinamide adenine dinucleotide/flavin adenine dinucleotide; NF-κB- nuclear factor κB; PDH- pyruvate dehydrogenase complex; PPARα- peroxisome proliferator-activated receptor-α; RAAS- renin-angiotensin-aldosterone system; ROS- reactive oxygen species; SERCA- sarcoplasmic reticulum calcium transport ATPase; SR- sarcoplasmic reticulum; TAG- triacylglycerol; TCA- tricarboxylic acid; O-GlcNAc- β-N-acetylglucosamine; UCP3- mitochondrial uncoupling protein 3.
(i) Hyperglycemia and hyperinsulinemia
Persistent hyperglycemia is considered to be a critical driver of DC, causing several metabolic, structural, and functional alterations. Hyperglycemia causes an increased utilization of glucose compared to fatty acids, with a subsequent increase in oxidative stress via generation of reactive oxygen species (ROS) from the mitochondria. Oxidative stress caused by the overproduction of superoxide in the mitochondrial respiratory chain leads to reduced myocardial contractility, and eventually induces myocardial fibrosis. ROS-mediated DNA damage, in turn activates DNA reparative enzymes, such as poly ADP ribose polymerase (PARP), which accelerates nicotinamide adenine dinucleotide (NAD+) catabolism and redirects glucose metabolism from its usual glycolytic pathway to an alternative pathway (Szabados et al., 1999; Pacher et al., 2002). Hyperglycemia is responsible for a series of cellular injuries mediated by increased hexosamine and polyol pathway flux, protein kinase C activation, and upregulated advanced glycation end product (AGE) levels (Perrone-Filardi et al., 2015).
Hyperglycemia results in increases in non-enzymatic glycation, oxidation of lipids and proteins, myocardial collagen and fibronectin production, cumulatively resulting in AGE accumulation and myocardial structural alterations (Davis et al., 2016). AGEs play a pivotal role in the progression of DC by stimulating collagen expression and accumulation, in addition to promoting collagen cross-linking, which leads to increased myocardial fibrosis and impaired passive relaxation (Marchetti, 2009). AGEs can also bind to their respective cell surface receptors, and upon doing so, promote maladaptive structural changes and impaired myocardial energetics. AGE accumulation also results in increased ROS production and activation of the transforming growth factor beta 1(TGF-β1)/SMAD pathway, as well as connective tissue production and fibrosis (Willemsen et al., 2012). Impaired glucose handling, as a result of hyperglycemia also contributes to maladaptive hexosamine biosynthesis pathway (HBP), resulting in dysregulation of β-N-acetylglucosamine (O-GlcNAc) in adipose and other non-cardiac tissues. Sustained maladaptive HBP and O-GlcNAc protein modification serves as a contributing mechanism of insulin resistance and progression of DC (Qin et al., 2017).
Hyperinsulinemia is another typical pathological feature of T2DM, secondary to insulin resistance, which is characterized by impaired myocardial insulin signaling (e.g. decreased Akt phosphorylation). Insulin resistance, which is associated with excessive energy substrates as a result of increased lipolysis, hepatic lipogenesis and gluconeogenesis, is characterized by the reduced uptake of glucose, which under normal physiological conditions can help buffer oxidative stress (Taegtmeyer et al., 2013). Compensatory hyperinsulinemia occurs as a result of decreased insulin sensitivity, leading to mitochondrial dysfunction, endoplasmic reticulum stress, impaired calcium homeostasis, activation of the sympathetic nervous system and the renin-angiotensin-aldosterone system, as well as a maladaptive immune response (Jia et al., 2016). The various genetic and epigenetic alterations that follow hyperinsulinemia, result in the activation of multiple transcription factors, which regulate extracellular and cellular protein expression, that contribute to diastolic dysfunction and HF (Wang and Hill, 2015).
(ii) Hyperlipidemia
Under physiological conditions, the heart mainly consumes fatty acids (FAs) as energy substrates (~70% of energy production) with glucose mainly accounting for the rest (Lopaschuk et al., 2010). The metabolic flexibility between FAs and glucose consumption is predominantly regulated by the Randle cycle and the cellular import of glucose and FAs via glucose transporter 4 (GLUT4) and CD36, respectively (Geraets et al., 2018). In DC patients, the balance between energy supply and demand is perturbed, with GLUT4 translocation to the cell surface being reduced, causing decreased glucose uptake, and increased CD36-mediated FA uptake. Increased circulating FAs and very-low-density lipoprotein-triacylglycerol concentrations result in an elevated lipid supply to the cardiomyocytes, which activates peroxisome proliferator-activated receptor (PPAR)-α, a nuclear receptor capable of activating transcripts for FA metabolism and shifting substrate utilization towards FAs (Schulze et al., 2016). However, the excessive lipid supply surpasses the mitochondrial oxidative capacity of cardiomyocytes, leading to cytosolic accumulation of lipid metabolites such as triacylglycerol (TAG), long-chain acyl coenzyme A (CoA), diacylglycerol (DAG), and ceramides, which can induce lipotoxicity. For instance, ceramides, the FAs non-oxidative pathway intermediates, are thought to interfere with cellular signaling, induce mitochondrial dysfunction, and apoptosis in cardiomyocytes, which eventually results in contractile dysfunction, contributing to DC and HFpEF (Summers, 2006). Moreover, increased FA oxidation (FAO) in the mitochondria is associated with augmented ROS production, resulting in the oxidation of cytoplasmic lipids into lipid peroxides (Boudina and Abel, 2006), which subsequently lead to the activation of mitochondrial uncoupling protein 3 (UCP3). This chain of events causes a reduction in mitochondrial membrane potential and uncoupling of mitochondrial oxidative metabolism, which perturbs ATP production (Rial et al., 2010).
Accelerated rates of FAO can also lead to increased production of acetyl CoA, which causes feedback inhibition of pyruvate dehydrogenase, but increases citrate levels via the Krebs cycle. The increase in citrate and ATP/ADP ratio decreases the enzymatic activity of phosphofructokinase and glucose metabolism, which subsequently reduces glucose oxidation, resulting in the development of myocardial insulin resistance (Martins et al., 2012). Recent studies (Song et al., 2013; Liu et al., 2015) have also shown that a striated muscle-specific E3 ligase, Mitsugumin 53 which was found to be overexpressed in an induced DC mouse model, not only destabilized the insulin receptor and insulin receptor substrate, but more importantly, upregulated PPAR-α and its target genes, resulting in lipid accumulation and lipotoxicity, thereby contributing to disease progression. Similarly, glycogen synthase kinase-3 alpha (GSK-3α) has also been found to mediate PPARα phosphorylation, a contributing factor in lipotoxic cardiomyopathy (Nakamura et al., 2019).
Cellular pathogenesis
A combination of the aforementioned metabolic derangements predisposes the diabetic heart to diastolic and/or systolic dysfunction, which is followed by structural remodeling such as LVH and myocardial fibrosis (Mizamtsidi et al., 2016). These structural and functional changes, as well as intracellular metabolic perturbations are mediated by several pathophysiological mechanisms that occur in cardiomyocytes. Although, the exact pathogenesis of DC has yet to be completely revealed, several possibilities have been proposed that can help explain the changes observed in cardiomyocytes.
(i) Mitochondrial injury and oxidative stress
Compared to other cell types, cardiomyocytes have the highest energy and oxygen demands, with the mitochondria serving as the main source of energy production. Any damage related to the mitochondria can therefore, render the myocardium functionally vulnerable (Schilling, 2015). In support of this notion, a close relationship has been reported between worsening myocardial contraction-relaxation properties and impaired mitochondrial function and dynamics (Montaigne et al., 2014).
Under conditions of excessive FAs and decreased metabolic flexibility, an upregulation of lipid metabolic pathways occurs, where activation of PPAR-α and PGC-1a/b help increase FAO, TAG synthesis, and mitochondrial biogenesis, in order to prevent generation of lipotoxic species, such as DAG and ceramides (Schilling, 2015). However, sustained activation of this metabolic pathway, together with increased rates of FAO and myocardial oxygen consumption can be detrimental. As FAO exceeds the capacity of downstream oxidative pathways, an uncoupling of FAO from oxidative phosphorylation (OXPHOS) has been reported to take place (Lemieux et al., 2011). Elevated mitochondrial FA flux results in ROS generation and upregulation of uncoupling proteins, both of which decrease mitochondrial efficiency and cause the accumulation of FAO metabolites, such as acylcarnitines and acetyl-CoA (Lopaschuk et al., 2010). With continued metabolic stress, further mitochondrial dysfunction and ROS generation worsens the imbalance between lipid uptake and oxidation, which overwhelms TAG synthesis pathways and promotes the generation of lipotoxic species (e.g. ceramides and DAG) (Park et al., 2008). Furthermore, excessive ROS production overwhelms the scavenger machinery, causing oxidative stress and together with lipid accumulation, triggers inflammation and cell death (Goldberg et al., 2012). Impairment of mitophagy is also reported to cause accumulation of ROS-generating mitochondria, thereby amplifying the detrimental effects of DC. (Kubli and Gustafsson, 2015).
ROS with accompanying oxidative stress play a detrimental role in damaging proteins, nucleic acids, and lipids, resulting in cardiomyocyte dysfunction and death (Arkat et al., 2016). ROS also facilitate cellular autophagy through the protein kinase C-mammalian target of rapamycin complex 1 (PKC-mTORC1) pathway (Inoguchi et al., 2003) and mediates apoptosis through the hexosamine pathway (Rajamani and Essop, 2010), by increasing intracellular calcium outflow. Importantly, ROS have a profound effect on the opening of the mitochondrial permeability transition pore (MPTP), resulting in the dissipation of the mitochondrial membrane potential (ΔΨm) and cellular calcium overload (Song et al., 2014).
Interestingly, cardiac tissue obtained from DC patients have been found to contain mitochondria with altered morphology, suggesting the possibility of impaired mitochondrial dynamics during disease progression (Kubli and Gustafsson, 2015). In support of this notion, hyperglycemia has been shown to induce mitochondrial fission via a dynamin-related protein 1 (DRP1)-dependent process (Hu et al., 2020) along with reduced ΔΨm and electron transport chain activity. However, despite activated fission and repressed fusion, only mitofusin 1 (MFN1) was found to be decreased, while mitofusin 2 (MFN2), fission 1 protein (FIS1) and optic atrophy protein 1 (OPA1) levels were unaltered in T2DM patient atrial tissue (Montaigne et al., 2014). Though it remains to be determined as to whether mitochondrial fusion/fission events are directly modulated by metabolic adaptations and cardiac function, studies have reported cardioprotective effects when targeting mitochondrial dynamics under pathological conditions (Ong et al., 2010; Ding et al., 2019).
(ii) Impaired calcium handling
Mitochondrial function is tightly regulated by calcium and hence, during increased workload, mitochondrial calcium uptake (via the mitochondrial calcium uniporter; MCU) is required to match energy supply to demand, in addition to preventing excessive production of ROS (Bertero and Maack, 2018). During the contraction phase, a small amount of calcium enters into the cytoplasm through L-type calcium channels, stimulating the sarcoplasmic reticulum (SR) to release calcium via ryanodine receptors and inositol triphosphate (IP3) receptors. During the relaxation phase, calcium is pumped back into the SR and extracellular space by sarcoplasmic reticulum calcium transport ATPase (SERCA) and sodium-calcium exchanger, respectively (Louch et al., 2015).
In DC, impaired cytosolic calcium handling subsequently hampers the MCU, causing energy depletion and oxidative stress (Duncan, 2011). Impaired calcium handling also interferes with excitation-contraction coupling, and together with lipid accumulation, increases oxidative stress, decreases ΔΨm and ATP depletion, and leads to the opening of the MPTP. This maladaptation of the mitochondria not only exacerbates the impaired calcium handling but it also initiates the release of pro-apoptotic factors, mainly cytochrome C, which activates the caspase signaling cascade, facilitating apoptotic cell death and cardiac remodeling (Kwong et al., 2014).
(iii) Inflammation and cardiomyocyte apoptosis
All of the changes reported above eventually result in inflammation and cardiomyocyte apoptosis. In support of this, studies have found several pro-inflammatory transcription factors (e.g. nuclear factor kappaB; NF-κB) and pro-inflammatory cytokines; including tumor necrosis factor-α (TNF-α), interleukins 6 and 8 (IL-6, IL-8), and monocyte chemotactic protein 1 (MCP-1) to be upregulated in the presence of DC, resulting in myocardial dysfunction, remodeling, and HF (Varga et al., 2015). Interestingly, NF-κB activation was found to be responsible for the induction of apoptosis, which led to a considerable number of apoptotic cardiomyocytes in the diabetic patient’s heart compared to a non-diabetic patient (Palomer et al., 2013).
Autophagy, which is the regulated removal of dysfunctional components of the cell, is a major homeostatic pathway. Perturbations in this pathway have also been observed in DC and speculated to have reciprocal causation with the initiation of apoptosis, however, the mechanism by which dysregulated autophagy leads to disease progression is debatable (Delbridge et al., 2017). For example, in a study involving T2DM patients with ischemic heart disease, apart from observing the activation of pro-apoptotic caspase-3, researchers found increased lipidated microtubule-associated protein 1A/1B-light chain 3 (LC3) and Beclin1 protein but with reduced p62 levels, thereby postulating the accumulation of autophagosomes (Munasinghe et al., 2016). Conversely, in another study, while LC3-II levels were found to be reduced, p62 levels were increased in DC mice. Interestingly, treatment with melatonin led to augmented autophagy with increased numbers of autophagosomes, which together with decreased apoptosis alleviated adverse left ventricular (LV) remodeling and improved cardiac function in DC (Delbridge et al., 2017; Zhang et al., 2017).
3. Clinical aspects of diabetic cardiomyopathy
Epidemiology of heart failure in diabetic patients
Epidemiological studies suggest that the risk of developing HF is up to 2.5 fold higher in patients with T2DM compared to age and sex-matched non-diabetic individuals (Kannel et al., 1974; Nichols et al., 2004). Even in patients with impaired glucose tolerance or insulin resistance, the risk of HF is 1.7 times increased when compared to non-diabetic individuals (Thrainsdottir et al., 2005). In the presence of T2DM, a 1% rise in glycated hemoglobin (HbA1c) levels has been found to be associated with an 8% increase in risk of HF (Iribarren et al., 2001; Seferovic and Paulus, 2015), while a 1% reduction in HbA1c levels correlated with a 16% reduction in risk of developing HF, linking poor diabetes control with increased propensity for HF (Stratton et al., 2000). Interestingly, T2DM was found to worsen the prognosis for patients with HFrEF, but even more so for those with HFpEF, with an increased risk of death and hospitalization (Sarma et al., 2013).
Patients with T2DM have a 40-75% higher risk of CVD or HF-related hospitalization compared to those without T2DM (Greenberg et al., 2007; Kristensen et al., 2017). In T2DM patients, the 1-year all-cause mortality in DM patients with HF is highly significant at 30%, which is approximately 1.5-fold greater than for HF patients without diabetes (Gustafsson et al., 2004). Moreover, in patients who are older than 65 years of age, the co-existence of HF has been shown to be associated with a 10-fold higher mortality risk (Bertoni et al., 2004). Similarly, patients with HF have been found to be associated with a three-fold higher prevalence of T2DM (10-20%), than patients without HF (3–6%) (Thrainsdottir et al., 2005; Lombardi et al., 2016). Thus, the epidemiological evidence suggests a bidirectional association between HF and T2DM in terms of incidence and prognosis, which supports the existence of DC as a clinical condition.
Clinical assessment of diabetic cardiomyopathy
Currently, there are no specific criteria for diagnosing DC, a condition that is believed to be asymptomatic for several years and only manifests when HF has been established (Lorenzo-Almoros et al., 2017; Jia et al., 2018).
(i) Biomarkers
A number of circulating biomarkers have been tested for their ability to predict clinical outcomes in diabetic patients with cardiac dysfunction but whether they are specific for DC is not clear. A prospective observational study of DM patients investigated the prognostic value of N-terminal pro-B-type natriuretic peptide (NT-proBNP), and found that low levels of NT-proBNP (<125 pg/mL) were associated with an ultra-low risk of short-term cardiovascular (CV) events (Huelsmann et al., 2008). It has also been reported in the I-Preserve (Irbesartan in Heart Failure With Preserved Systolic Function) randomized trial that HFpEF patients with diabetes have a higher median NT-proBNP concentration (403 versus 320 pg/mL; P<0.01) with poorer prognosis (Kristensen et al., 2017). However, in another study, while B-type natriuretic protein (BNP) levels were found to be elevated in asymptomatic DM patients with LVH, it was not sufficiently sensitive to identify subclinical cardiac dysfunction. This finding was supported by another study, where T2DM patients without hypertension or coronary atherosclerosis showed deterioration of LV longitudinal shortening, but with no significant difference in BNP levels (Enomoto et al., 2015). Furthermore, in DM patients without known CVD, measurement of inflammation or apoptotic makers such as C-reactive protein or fibrinogen have been reported to provide minor incremental value to current risk assessment (Cosentino et al., 2019). Several other potential biomarkers for diabetes-induced impairment of myocardial function have also been identified, including circulating microRNAs (Halushka et al., 2019), fibrotic markers (e.g., TGFβ) (Asbun and Villarreal, 2006) and glucose metabolites (e.g., O-GlcNAc) that can be detected in circulating erythrocytes (Marwick et al., 2018).
In summary, there is currently no consensus on the biomarker profile that can be used to screen for DC and to monitor the progression of DC before its clinical manifestation of HF. Therefore, detection of early and reversible events such as cardiac hypertrophy and contractibility using imaging may be more helpful.
(ii) Echocardiography
Echocardiography is the favored imaging modality for evaluating structural and functional cardiac abnormalities associated with DM (Yancy et al., 2017; Cosentino et al., 2019). It provides accurate identification of the structural deformations observed during the early stages of DC, such as increased LV mass, LVH (Pham et al., 2015), diastolic dysfunction, and impaired LV deformation. An evaluation of two-dimensional (2D) echocardiography images obtained from two prospective T2DM patient cohorts found that those with the lowest LV mass index, and transmitral early diastolic rapid filling velocity to atrial contraction late filling velocity (E/A) ratio, had the highest LV ejection fraction, and the better prognosis in terms of fewer deaths and CV-related hospitalizations, compared to those with higher LV mass and advanced LV systolic and diastolic dysfunction (Patil et al., 2011; Ernande et al., 2017). Consistent with these findings, another study found that echocardiographic abnormalities were associated with decreased quality of life and poorer prognosis in DC patients (Kristensen et al., 2017). Furthermore, 2D echocardiography can also be used to detect epicardial adipose tissue accumulation, which has been shown to correlate with HF and associated biomarkers in DC (Lima-Martínez et al., 2016).
More recently, echocardiography tissue Doppler imaging (TDI) has been tested for monitoring myocardial mechanical deformations. The early transmitral velocity to TDI mitral annular early diastolic velocity (E/E’) ratio, strain rate, and global and regional ventricular strains have been described as practical measures of diastolic dysfunction, which characterize the early-stage of DC (Di Bonito et al., 2005; Seferovic and Paulus, 2015). In addition, speckle tracking echocardiography, which is a three dimensional (3D) technique, has been used to establish a correlation between DM microangiopathy and subclinical LV dysfunction by impaired longitudinal shortening and circumferential strains (Enomoto et al., 2016).
In summary, although echocardiography can be used to detect the early manifestations of DC in terms of the presence of increased LV mass, LVH, and diastolic dysfunction, there exist no specific criteria for diagnosing DC by echocardiography.
(iii) Cardiovascular magnetic resonance imaging
Cardiovascular magnetic resonance (CMR) imaging may also play an important role in the diagnosis of DC. In comparison to echocardiography, the key advantage of CMR is the ability to characterize the myocardium in terms of myocardial edema and inflammation (by T2-mapping), the presence of interstitial fibrosis (by T1-mapping), or myocardial infarction (by late gadolinium enhancement [LGE] imaging, allowing one to exclude other causes of cardiomyopathy such as ischemic cardiomyopathy. Late CMR and myocardial tagging CMR (to assess LV stain) in DM patients have been used to evaluate the presence of ischemic and non-ischemic scar, as well as circumferential strain, which in combination can be used to image myocardial interstitial fibrosis as a marker of DC (Armstrong et al., 2017). In the MESA (Multi-Ethnic Study of Atherosclerosis) study, tagged CMR demonstrated a correlation between impaired glucose tolerance and higher LV mass index, increased LV mass-to-volume ratio, and lower longitudinal and circumferential shortening, which was indicative of LV concentric remodeling and systolic dysfunction (Yoneyama et al., 2018).
Another key advantage of CMR, is its ability to detect the metabolic changes that take place during the early stages of DC (Scheuermann-Freestone et al., 2003). Blood-oxygen level-dependent (BOLD) CMR and stress perfusion CMR have been used to assess the myocardial energetic performance of T2DM patients under increased workload, where it determined that the cardiac energetic deficit in DC was exacerbated with exercise, as evidenced by reduced phosphocreatine to adenosine triphosphate ratios (PCr/ATP) and impaired myocardial perfusion reserve index (Levelt et al., 2016).
In summary, compared with echocardiography, CMR can provide unique insights into the pathophysiology underlying DC (such as interstitial fibrosis, inflammation, and metabolic perturbations), but again there exist no specific criteria for diagnosing DC by CMR.
Clinical management of diabetic cardiomyopathy
In accordance with the identified pathophysiology involved in DC progression, various therapeutic agents have been shown to delay the deterioration of myocardial function in patients with DM, thereby improving the prognosis of DC patients.
(i) Glucose-lowering agents
Since hyperglycemia is widely acknowledged as the primary cause that drives the progression of DC, efforts to tightly control glucose concentrations seem a logical strategy. Metformin, an activator of adenosine monophosphate-activated protein kinase (AMPK) has been considered as a first-line treatment for T2DM as it has been shown to reduce the risk and improve the clinical course of HF (Packer, 2018). In a systematic review of observational studies, metformin was identified as a safe glucose-lowering agent for patients with T2DM and HF, as evidenced by reduced mortality (approximately 15%), and is even appropriate for patients who have progressed to HFrEF and chronic kidney disease (Eurich et al., 2013). However, a meta-analysis of 13 randomized controlled trials revealed metformin to have no significant improvement on CV mortality and HF risk ratio (Boussageon et al., 2012). Moreover, in a recent Phase IV clinical trial involving T2DM patients, no significant changes in LV mass index, BNP levels or E/E’ were observed in the metformin-treated group at 1 year follow-up (Ono et al., 2019). The proposed benefits of metformin in treating DM and HF therefore remain controversial.
Sulphonylureas and insulin are also recommended second- and third-line therapies, but their safety in HF is ambiguous (Seferovic et al., 2018). Alternatively, thiazolidinediones (TZDs), which are PPAR-gamma (PPARγ) agonists, have been shown to reverse metabolic dysfunction and improve insulin resistance associated with T2DM (Gross and Staels, 2007), however, the TZD, Rosiglitazone, was shown to be associated with increased CV risk, especially for HF events (Wallach et al., 2020).
The newer incretin-based anti-diabetic agent, glucagon-like peptide-1 receptor (GLP-1R) agonist has also been used for DC treatment. Liraglutide, a typical GLP-1R agonist, which was reported to impart beneficial effects in DM and HF patients, has been tested in several clinical trials, albeit with controversial outcomes. In the LEADER (Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results) study (Marso et al., 2016), liraglutide was found to moderate the risk of CV events and death in T2DM patients. Conversely, in another study, which focused on South Asian ethnic groups, 26-weeks of liraglutide treatment was found to have no effect on myocardial function (Paiman et al., 2019). Moreover, in the LIVE (The Effect of Liraglutide on Left Ventricular Function in Chronic Heart Failure Patients With and Without Type 2 Diabetes Mellitus) and FIGHT (Functional Impact of GLP-1 for Heart Failure Treatment) studies, liraglutide failed to enhance LV systolic function and did not improve clinical stability in HF patients with or without diabetes (Margulies et al., 2016; Jorsal et al., 2017). Despite these controversial outcomes, a recent systematic review that included 7 clinical trials reported that GLP-1R agonist treatment exerted beneficial effects by reducing major adverse CV events by 12% and hospitalization for HF by 9% among T2DM patients (Kristensen et al., 2019).
In light of these outcomes, it may seem that the tight control of glucose may not be the ideal strategy for reducing CV risk in T2DM. In support, a meta-analysis of prospective randomized controlled trials published over a period of 20 years failed to identify robust CV benefits when HbA1c levels were below 7.0% (Wang et al., 2015).
More recently, sodium-glucose transporter 2 (SGLT2) inhibitors, such as Empagliflozin, Canagliflozin, and Dapagliflozin, have been identified to have glycemic effects in T2DM with accompanying multi-system health benefits, especially cardioprotection (Vaduganathan and Butler, 2019). In the EMPA-REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) study, Empagliflozin was associated with a 35% reduction in the risk of hospitalization for HF and a 38% decrease in CV-related death in T2DM patients (Zinman et al., 2015). Similarly, the CANVAS (Canagliflozin Cardiovascular Assessment Study) and CANVAS-R (A Study of the Effects of Canagliflozin on Renal Endpoints in Adult Participants With Type 2 Diabetes Mellitus) studies (Neal et al., 2017), which included a longer follow-up period reported Canagliflozin alleviated the risk of CV events in T2DM patients. Moreover, in the CVD-REAL (Comparative Effectiveness of Cardiovascular Outcomes in New Users of SGLT-2 Inhibitors) study (which included all 3 SGLT2 inhibitors), when compared to other glucose-lowering agents, treatment with SGLT2 inhibitors was found to lower the risk of hospitalization for HF and CV-related death (Kosiborod et al., 2017). Recently, in a large multicenter randomized clinical trial (DAPA-HF (Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure)) involving HFrEF patients, Dapagliflozin was found to successfully reduce the risk of worsening HF or death from CV-related causes, regardless of the presence or absence of diabetes (McMurray et al., 2019). Imaging studies have also supported the beneficial effects of Dapagliflozin. In a prospective multicenter study, 6-month Dapagliflozin treatment was found to improve LV diastolic function in T2DM patients with HF comorbidities as evidenced by significant decrease in E/E’, LV mass index, and left atrial volume index (Soga et al., 2018). When considering whether SGLT2 inhibitors can have a more pronounced effect on HFpEF (as opposed to HFrEF), initial studies suggest that there is no such preference, as further analysis of the CANVAS study (Figtree et al., 2019) revealed that Canagliflozin had no clear difference in risk-reducing effects between HFrEF and assumed HFpEF events. However, dedicated HFpEF trials are needed to confirm these observations (Ejiri et al., 2019).
In light of these positive outcomes associated with SGLT2 inhibition, researchers have attempted to elucidate its mode of action. In an angiotensin-II induced DC mouse model, one month of Dapagliflozin treatment was reported to attenuate myocardial fibrosis and inflammation, resulting in an increase in LV ejection fraction, which was speculated to be due to reduction of intracellular calcium transients and decreased production of ROS (Arow et al., 2020). Furthermore, while one study in HF suggested that the cardiovascular protective effect of Empagliflozin is mediated by blockade of the sodium-hydrogen exchanger and stimulation of a signaling cascade that attenuates cell death (Iborra-Egea et al., 2019), another study implies the switching of myocardial fuel utilization from glucose to ketone bodies (Santos-Gallego et al., 2019a). In summary, given the successful outcomes of the clinical trials and associated reduction in risk of hospitalization of HF patients with or without T2DM (Seferovic et al., 2020), SGLT-2 inhibitors have the potential to treat both DC and HF in the near future, although further studies are needed to understanding their mode of action (Jensen et al., 2019) and assess their cardiac benefit in HF (Santos-Gallego et al., 2019b).
(ii) Lipid-lowering agents
Dyslipidemia, which consists of high plasma triglycerides and low-density lipoproteins, is another major risk factor for DC. Altered hepatic lipid metabolism, adipocyte insulin resistance, and increased FA flux can trigger maladaptive myocardial FA metabolism. Statins are considered conventional lipid-lowering agents (Rogers et al., 2014) and play an important role in reducing circulating lipids and intra-myocardial inflammation (Antonopoulos et al., 2012). In an early meta-analysis study, which includes 14 randomized trials and 17,220 T2DM patients, statin therapy was reported to significantly reduce vascular-related mortality and major vascular event rates, irrespective of any other baseline characteristic (Doll, 2008). In the TOPCAT (Aldosterone Antagonist Therapy for Adults With Heart Failure and Preserved Systolic Function) study, statin therapy was associated with improved all-cause death and CV mortality in HFpEF patients (Tsujimoto and Kajio, 2018). Whether statin therapy can prevent the occurrence and progression of DC in DM patients is not known and remains to be tested (Liberale et al., 2019).
Fibrates, which are small molecule ligands of PPARα are also considered to have lipid-lowering properties by upregulating FA metabolism and reducing circulating triglyceride levels (Nakamura and Sadoshima, 2019). Fenofibrate therapy in combination with statins was shown to reduce CV mortality and the rate of fatal and non-fatal chronic HF in T2DM patients who are at high CV risk (Papademetriou et al., 2017). In light of these findings, a selective PPARα modulator, Pemafibrate is being tested in an on-going clinical trial (PROMINENT (Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients With Diabetes)) aimed at assessing the remaining residual CV risk following treatment to reduce low-density lipoprotein cholesterol in T2DM individuals with dyslipidemia (Pradhan et al., 2018).
Omega-3 fatty acids, which are able to reduce triglycerides and non-high-density lipoprotein levels have also been reported to have cardio-metabolic benefits (Burke et al., 2017). When patients with chronic HF were supplemented with omega-3 polyunsaturated FA (Omega-3 PUFA), improvements in left diastolic function and decreased BNP levels were observed during a 6-month follow-up (Chrysohoou et al., 2016). Conversely, in a larger double-blind study, omega-3 PUFA supplementation was found to have no effect in patients with high risk CV events and impaired fasting glucose, impaired glucose tolerance, or diabetes during a median follow up of 6-years (Investigators et al., 2012).
(iii) Other agents
The renin-angiotensin-aldosterone system (RAAS) and neprilysin inhibitors (Bernardi et al., 2016) and β-blockers (Ibrahim et al., 2019) are alternative agents recommended in the 2019 ESC Guidelines (Cosentino et al., 2019) for improving the prognosis and reducing all-cause death and hospitalization of HFrEF patients with diabetes. In the PARADIGM-HF (Study to Evaluate the Efficacy and Safety of LCZ696 Compared to Enalapril on Morbidity and Mortality of Patients With Chronic Heart Failure) study, which involved HFrEF patients with high HbA1c levels, LCZ696 (Sacubitril and Valsartan, RAAS and neprilysin inhibitors) was found to have beneficial effects when compared with Enalapril (Kristensen et al., 2016). A meta-analysis of large-scale clinical trials has also revealed beta-blocker therapy (Carvedilol or Metoprolol) to be beneficial in chronic HF patients with DM, albeit with a lesser magnitude than those without DM (Haas et al., 2003).
In addition to these compounds, other potential therapeutic options for DC patient management have also been identified. For example, a phosphodiesterase-5 inhibitor was reported to control disease progression in DC patients by reducing circulating and cellular levels of interleukin 8 (a pro-inflammatory chemokine) (Matyas et al., 2017; Giannattasio et al., 2019). miRNAs present in exosomes on the other hand have been shown to regulate apoptosis, inflammation, fibrosis, and angiogenesis (Jung et al., 2017; Nandi and Mishra, 2018). Finally, vitamin D has been reported to improve CV outcomes in DM by regulating AGE signaling (Lee et al., 2019). In light of these promising initial outcomes, further evaluation of these compounds is warranted.
4. Disease models
Animal models
Animal models (rodents in particular), continue to represent the most commonly used and successful approach towards addressing a biological question (Jimenez-Tellez and Greenway, 2019). The first animal model (Zucker rats) reported for their use in diabetes-related studies can be traced back to the 1960s (Zucker and Zucker, 1962), and is still widely being used to investigate the pathogenesis and treatment efficacy for diabetes-related complications.
(i) Spontaneous DC model
The spontaneous DC model that lacks artificial manipulation, is generated in the presence of natural genetic mutations coupled with long time breeding. Due to similar myocardial pathophysiological phenotypes, ob/ob mice (Zhang et al., 1994), db/db mice (Coleman, 1978), and Zucker diabetic fatty rats (Iida et al., 1996) are often used in studies investigating diabetes-induced CVD. For example, ob/ob mice develop diabetes as a consequence of recessive mutations in the obesity gene, which also cause a reduction in hypothalamic leptin action. After 15 weeks, these mice exhibit increased FAO rates and myocardial triglyceride storage, as well as decreased glucose oxidation rates accompanied by cardiac insulin resistance and diastolic dysfunction. Db/db mice develop a similar phenotype as ob/ob mice but at a much faster rate (8 weeks). While these models share many similar traits with the hearts of T2DM patients, leptin deficiency can induce several compounding factors when attempting to identify pathological mechanisms (Bugger and Abel, 2009).
(ii) Induced DC model
To circumvent potential issues surrounding altered leptin signaling that occurs in spontaneous models, researchers have shifted their focus towards high fat diet- or streptozotocin-induced DC models. For instance, when Wistar rats were supplemented with a high-fat diet (containing 25 wt% fat, 32 wt% protein and 25 wt% carbohydrate, as well as more palmitate and oleate) for 7 weeks, their blood glucose levels and heart weight were found to be increased, similar to a hypertrophic cardiac phenotype. Consistently, these rats exhibited a higher basal contractile force, impaired recovery from increased workload, in addition to decreased insulin-mediated protection against calcium overload. The excessive dietary fat was found to induce myocardial insulin resistance and was also linked to the reduction in phospholamban phosphorylation, leading to cardiac functional and structural changes observed in DC (Ouwens et al., 2005).
(iii) Gene-edited DC model
With ever-growing interest for assessing the genetic influence on pathological mechanisms, together with the increase in demand for targeted therapies, genetically engineered mice are being routinely used to evaluate the specific role of discreet pathways in the development of diabetes-related cardiac dysfunction. In a recent study, mutations in seipin (encoded by BSCL2), an endoplasmic reticulum transmembrane protein, were found to cause severe lipodystrophy, resulting in diabetes. Seipin knockout (SKO) mice have also been generated by a two-step process, whereby researchers first introduced loxP sites on either side of exons 5-7 of BSCL2. When crossed with Cre transgenic mice, exons 5-7 were removed and the resultant SKO model was generated (McIlroy et al., 2018). Adult SKO mice were found to display glucose intolerance and reduced insulin sensitivity, in addition to LV concentric hypertrophy and diastolic dysfunction; pathological features resembling the major cardiac structural and functional abnormalities of lean DM HFpEF patients in Asia. Furthermore, SKO mice were found to have increased interstitial fibrosis and altered titin phosphorylation, which can be potential therapeutic targets for preventing diabetes-related HFpEF (Bai et al., 2019).
Cellular models
Establishing an ideal animal model that accurately mimics DC is a critical prerequisite for identifying underlying mechanisms of the disease (Bugger and Abel, 2009). Despite a certain degree of success, animal models are still being questioned with regard to their inability to recapitulate the complete phenotype of DC. For instance, after more than 18 weeks (and 3 doses of streptozotocin), diabetic mice fail to exhibit cardiomyocyte hypertrophy and increased LV mass, which are common features in DC patients. Moreover, most animal-related studies are limited to males; whereas in humans, women are more vulnerable to T2DM (Tate et al., 2019). Inter-species differences are another factor, which can complicate DC research by hampering the translation of newly identified therapies for patient management.
To gain better insight into the cellular pathogenesis of DC and to bypass species differences, in vitro human cellular models can be considered. However, limited availability of heart-tissue during the early stages of disease development and ethical concerns surrounding human embryonic stem cells (Thomson et al., 1998), make it nearly impossible to study human material. Fortunately, with the discovery of human induced pluripotent stem cells (hiPSCs) (Takahashi et al., 2007), researchers now have access to an unlimited source of human cardiomyocytes (hiPSC-CMs). Importantly, since hiPSCs contain the complete genetic information of the individual from which they were derived, patient-specific cardiomyocytes can be generated for modeling diseases in a petri dish. Currently, there are numerous protocols available for differentiating hiPSCs into cardiomyocytes (Lian et al., 2012; Burridge et al., 2014; Mehta et al., 2014a), and with these advances, researchers have gone on to model various cardiac diseases, including Long QT syndrome (Moretti et al., 2010; Mehta et al., 2014b), hypertrophic cardiomyopathy (Lan et al., 2013; Viswanathan et al., 2018), dilated cardiomyopathy (Sun et al., 2012), and other disorders (Huang et al., 2011; Kim et al., 2013; Wang et al., 2014). Apart from their self-renewal ability, another advantage of using hiPSCs is that they can be generated from a wide variety of somatic cell types, which include but are not limited to dermal fibroblasts, peripheral blood mononuclear cells and T-cells; cell types which are relatively easy to obtain through minimal-invasive procedures (Musunuru et al., 2018).
Using iPSC technology, a model of DC has been established (Drawnel et al., 2014). In this study, researchers first attempted to induce a DC phenotype in healthy hiPSC-CMs, by modulating FA, glucose, and insulin concentrations in culture. Upon being exposed to this diabetic environment for 48 hours, hiPSC-CMs displayed hypertrophy and structural abnormalities, including loss in sarcomere integrity and myofilament disarray. The study also focused on generating hiPSCs from two T2DM patients with varied disease severity. Interestingly, these hiPSC-CMs did not require the diabetic environment to manifest the cellular abnormalities, as they spontaneously gave rise to the diseased phenotype, in addition to recapitulating the severity of the original clinical status. Collectively, such models can potentially be used to identify inherent genetic links and cellular mechanisms underlying DC (Figure 2).
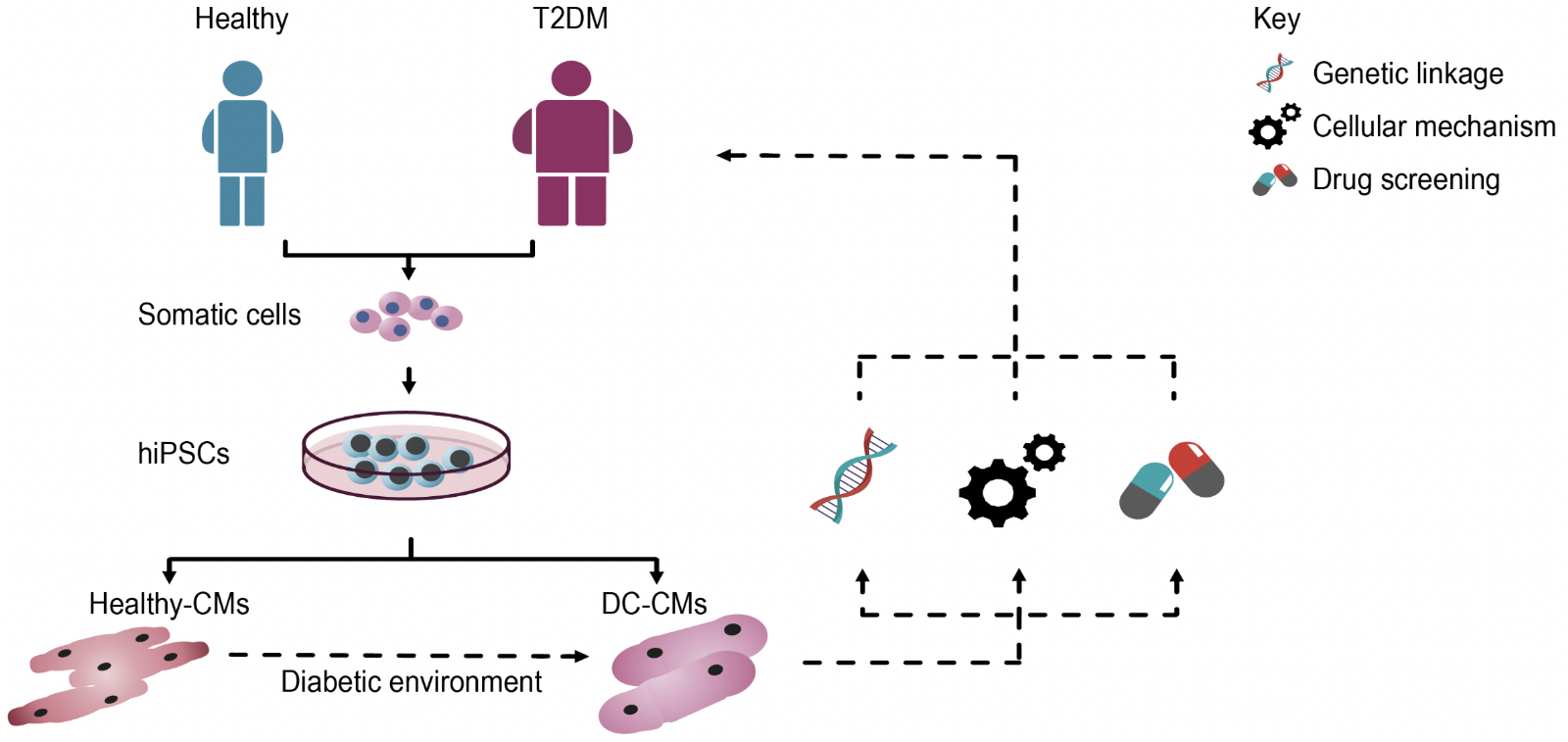
In a new window | Download PPT
Figure 2: Schematic representation illustrating the use of hiPSCs to model DC and for identifying novel targets for patient-specific therapies. Initially, somatic cells (e.g. dermal fibroblasts, PBMCs) isolated from patients with T2DM are reprogrammed into hiPSCs, which are then differentiated into functional cardiomyocytes (hiPSC-CMs), which spontaneously recapitulate the disease phenotype. Alternatively, hiPSC-CMs derived from healthy subjects can be exposed to a diabetic culture environment to induce a disease phenotype. These hiPSC-CMs can then be used to investigate genetic links and cellular mechanisms underlying DC, as well as serve as a drug screening platform to identify novel compounds for patient-specific therapies.
As a drug screening platform, hiPSC-CMs have also been used to gain insight into the mechanistic action of empagliflozin, which was found to reduce HF and sudden cardiac death (Zinman et al., 2015). When hiPSC-CMs were cultured in a diabetic-like environment (insulin-free and high-glucose), researchers observed cellular hypertrophy, reduced contractility, and calcium-handling abnormalities. Consistent with the clinical study, treatment with empagliflozin effectively reduced cardiac abnormalities, without altering the viability or glycolytic capacity of hiPSC-CMs (Ng et al., 2018). Notably, this study showed that empagliflozin probably exerted its effects via the down-regulation of SGLT1, SGLT2, and GLUT1, which serves as a plausible explanation for the beneficial effects observed in the EMPA-REG trial (Zinman et al., 2015).
Acylcarnitines, which are mainly derived through incomplete FAO, are commonly used as a biomarker profile to assess metabolic derangements associated with insulin resistance and T2DM (Schooneman et al., 2013; Sun et al., 2016). They have also been reported to significantly improve predictability of T2DM, independent of conventional risk factors (Sun et al., 2016). The reason for acylcarnitine accumulation during insulin resistance however, remains controversial. Initially, it was thought that cytosolic long-chain FA accumulation was a result of mitochondrial dysfunction and decreased FAO (Morino et al., 2006), however, in skeletal muscle obtained from Zucker diabetic fatty rats, FAO was found to be increased with high levels of acylcarnitines (Koves et al., 2008). Interestingly, a depletion in TCA cycle intermediates was also observed in this study, which suggested a mismatch between FAO and TCA flux under conditions of lipid-induced insulin-resistance. Similar to acylcarnitines, specific amino acid profiles have also been reported to predict future incidences of diabetes and could therefore aid in diabetes risk assessment (Wang et al., 2011). Considering that acylcarnitines can be derived from amino acids, an increase in branched-chain amino acids was found to be associated with increased short-chain acylcarnitines and insulin-resistance (Newgard et al., 2009). In summary, though several studies have associated accumulation of acylcarnitines with insulin-resistance, it is unclear as to whether acylcarnitines are indicators of insulin resistance or primary contributors to the diseased state. Previously it has been shown that changes in acylcarnitines, TCA cycle intermediates and amino acids can be documented in healthy hiPSC-CMs supplemented with FA (Ramachandra et al., 2018). Similar metabolomic profiling of diseased hiPSC-CMs, coupled with assays that measure oxygen consumption rates (e.g. Seahorse Biosciences) could help establish the link between metabolite imbalance, metabolic derangement, and mitochondrial dysfunction in addition to identifying important mechanisms that underlie impaired lipid metabolism, insulin resistance, and DC.
Limitations of hiPSC-CMs
Although hiPSC-CMs have several advantages, they do possess some limitations. The purity and maturity of cardiomyocytes is a major concern where researchers continue to direct their efforts. In order to improve the purity of hiPSC-CMs, researchers resort to manual enrichment, fluorescence sorting, metabolic, or drug selection. Taking advantage of the metabolic plasticity of cardiomyocytes, when hiPSC derivatives were cultured in glucose-depleted, but lactate-rich culture conditions for 20 days, a highly pure (99%) hiPSC-CM population can be obtained (Tohyama et al., 2013). Similarly, when an antibiotic marker (neomycin) was inserted under the control of the myosin light chain 2 promoter, a highly pure (98%) ventricular hiPSC-CM population can be obtained under drug selection (Li et al., 2017). While further work is needed to determine whether these methods of selection alter the normal physiology of hiPSC-CMs, cell-sorting based on mitochondrial content, which takes advantage of the abundant mitochondria present in cardiomyocytes, could be an alternative strategy for improving purity (Li et al., 2019).
Apart from purity, the maturity of hiPSC-CMs is considered to be a more important and challenging aspect that needs to be addressed. When modeling DC, it is essential for hiPSC-CMs to recapitulate the metabolic perturbations that manifest with the disease, but evidence which suggests hiPSC-CMs to be fetal-like, weakens their credibility for modeling adult CV diseases (Bowman et al., 2019). A number of strategies have been attempted to enhance the maturity of hiPSC-CMs, including simple methods such as prolonged culture (200 days) (Ebert et al., 2019), where metabolic preference were found to shift from glycolytic to OXPHOS and FAO, in comparison to short-term cultures (day 30). To circumvent time-related issues with prolonged cultures, direct supplementation of FA has been shown to advance the metabolic phenotype in 30 days (Ramachandra et al., 2018). The supplementation of culture conditions with physiological postnatal factors also seems to be gaining interest. Consistently, researchers have shown that the introduction of thyroid and glucocorticoid hormones during the differentiation process, coupled with further modulation on matrices mimicking physiological stiffness, can induce the maturation of several components, including T-tubule development, enhanced calcium-induced calcium release, and more ventricular-like excitation-contraction coupling (Parikh et al., 2017). Apart from manipulating culture conditions, approaches that modulate physical properties have also been implemented to accelerate the maturation of hiPSC-CMs. For instance, when a collagen-based bioengineered human cardiac tissue that had been generated from hiPSC-CMs was subjected to static mechanical stress and electrical pacing, researchers were able to attain structural maturation with improved mechanical features and increased force generation (Ruan et al., 2016). Although further work is needed to convincingly show that hiPSC-CMs can be manipulated to closely mimic their adult counterparts, current strategies have significantly contributed towards enhancing maturation over the past few years, highlighting the capabilities and feasibility of hiPSC-CMs for modeling metabolic perturbations in DC.
5. Conclusion
Though first described over 40 years ago, DC still remains a severe medical complication associated with considerably high mortality and morbidity in DM patients. The disease is characterized by molecular, metabolic, structural, and functional changes not only in the heart, but throughout the whole body. While several mechanisms such as chronic hyperglycemia, hyperlipidemia, insulin resistance, mitochondrial damage, oxidative stress, and inflammation have been proposed, clear, feasible, diagnostic criteria and specific therapeutic approaches have yet to be identified. Given the importance of metabolism and mitochondrial bioenergetics in normal cardiac physiology, maladaptive metabolic alterations with subsequent mitochondrial dysfunction can cause energy deficiency, resulting in DC, thereby emphasizing the need for robust cellular models that can recapitulate these pathological processes. Modeling the DC phenotype with hiPSC-CMs can provide an unprecedented opportunity for detailed observation into disease manifestation and progression, with the ultimate aim of identifying novel targets for better patient management strategies.
Conflicts and disclosures
All authors have no conflicts or disclosures.
Acknowledgement
Jonathan Yap is supported by National Institutes of Health grant F31 HL139082. Shuo Cong is supported by Fudan University Shanghai Medical College under its Exchange Program Scholarship for Postgraduate Student (2019201). Chrishan Ramachandra is supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (OF-YIRG) – (NMRC/OFYIRG/0073/2018) and through the National Health Innovation Centre Singapore under its Innovation to Develop Grant (NHIC-I2S-1811007). Lai Wei is supported by the National Nature Science Foundation of China (81974272). Derek Hausenloy is supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
References
Shuo Cong1,2
1Department of Cardiac Surgery, Zhongshan Hospital, Fudan University, Shanghai, China. 2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
Chrishan J.A. Ramachandra2,3
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 3Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore.
KP Myu Mai Ja2
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore.
Jonathan Yap4
4Center for Cardiovascular Research, John A. Burns School of Medicine, University of Hawaii, USA.
Winston Shim5
5Health and Social Sciences Cluster, Singapore Institute of Technology, Singapore.
Lai Wei1,6
1Department of Cardiac Surgery, Zhongshan Hospital, Fudan University, Shanghai, China. 6Shanghai Engineering Research Centre of Cardiac Valve, Shanghai, China.
Derek J. Hausenloy2,3,7–9
2National Heart Research Institute Singapore, National Heart Centre Singapore, Singapore. 3Cardiovascular and Metabolic Disorders Programme, Duke-NUS Medical School, Singapore. 7Yong Loo Lin Medical School, National University of Singapore, Singapore. 8The Hatter Cardiovascular Institute, University College London, London, UK. 9Cardiovascular Research Centre, College of Medical and Health Sciences, Asia University, Taiwan.
Shuo Cong, Chrishan J.A. Ramachandra and KP Myu Mai Ja contributed equally to this article.
Corresponding author:
Derek J. Hausenloy
Email: derek.hausenloy@duke-nus.edu.sg
In a new window | Download PPT
Figure 1: Schematic illustration of cardiac metabolic alterations in DC. In T2DM, hyperglycemia and hyperlipidemia cause a series of changes within the myocardium, resulting in decreased glucose uptake but with concomitant increase in FAO. Excessive FA metabolites and non-glycolytic glucose products mediate lipotoxicity and glucotoxity, respectively. As a result of maladaptive metabolism, increased intracellular ROS activates mitochondrial uncoupling, making ATP production less efficient, and also causes opening of the mPTP, thereby initiating apoptosis. Abbreviations: AGE- advanced glycation end product; CPT- carnitine palmitoyltransferase; CoA- coenzyme A; FACS- fatty acyl-coenzyme A synthetase; CD36- fatty acid translocase; DAG- diacylglycerol; ETC- electron Transport Chain; FA- fatty acid; GLUT4- glucose transporters 4; GSK-3α- glycogen synthase kinase-3α; IMS- inner membrane space; MG 53- mitsugumin 53; mPTP- mitochondrial permeability transition pore; MCU- mitochondrial calcium uniporter; NAD+- nicotinamide adenine dinucleotide; NADH/FADH2- nicotinamide adenine dinucleotide/flavin adenine dinucleotide; NF-κB- nuclear factor κB; PDH- pyruvate dehydrogenase complex; PPARα- peroxisome proliferator-activated receptor-α; RAAS- renin-angiotensin-aldosterone system; ROS- reactive oxygen species; SERCA- sarcoplasmic reticulum calcium transport ATPase; SR- sarcoplasmic reticulum; TAG- triacylglycerol; TCA- tricarboxylic acid; O-GlcNAc- β-N-acetylglucosamine; UCP3- mitochondrial uncoupling protein 3.
In a new window | Download PPT
Figure 2: Schematic representation illustrating the use of hiPSCs to model DC and for identifying novel targets for patient-specific therapies. Initially, somatic cells (e.g. dermal fibroblasts, PBMCs) isolated from patients with T2DM are reprogrammed into hiPSCs, which are then differentiated into functional cardiomyocytes (hiPSC-CMs), which spontaneously recapitulate the disease phenotype. Alternatively, hiPSC-CMs derived from healthy subjects can be exposed to a diabetic culture environment to induce a disease phenotype. These hiPSC-CMs can then be used to investigate genetic links and cellular mechanisms underlying DC, as well as serve as a drug screening platform to identify novel compounds for patient-specific therapies.
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 11550 | 32 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA