Browse Articles
Conditioning Medicine
International bi-monthly journal of cell signaling, tissue protection, and translational research.
Models of conditioning in neuromuscular junction as possible strategy of repair in spinal muscular atrophy
Time:2024-11-20
Number:4955
Author Affiliations

Conditioning Medicine 2024. 7(1): 15-21.
Abstract
Preconditioning is a phenomenon wherein a sub-lethal insult induces cellular and tissue resistance to a later severe injury that can aid in counteracting neurodegeneration in different tissues, including the brain. Although preconditioning is widely investigated under various pathological conditions, such as stroke, little is known about its role in spinal muscular atrophy (SMA). SMA is a neurodegenerative disease caused by the lack of survival motor neuron (SMN) protein. In recent years, treatments able to restore SMN protein levels have been developed. However, these therapeutic strategies are not a definitive cure, and molecular mechanisms that lead to SMA are not completely understood. In this context, further studies to elucidate the molecular basis of the disease and the development of complementary strategies to regenerate damaged motor axons could play a pivotal role in ameliorating the outcome of SMA. In this regard, it has been recently demonstrated that the myelination of motor axons is impaired in SMA, and boosting neuregulins triggers the ensheathment of axons. This review discusses how a “preconditioning” phenomenon could trigger axonal terminal regeneration. We examine the “preconditioning stimuli” that can ameliorate this aspect of SMA pathology, which cannot be reversed by postnatal SMN protein induction.
Keywords: Spinal muscular atrophy, Neuromuscular junction regeneration, Schwann cells, Myelination
Abstract
Preconditioning is a phenomenon wherein a sub-lethal insult induces cellular and tissue resistance to a later severe injury that can aid in counteracting neurodegeneration in different tissues, including the brain. Although preconditioning is widely investigated under various pathological conditions, such as stroke, little is known about its role in spinal muscular atrophy (SMA). SMA is a neurodegenerative disease caused by the lack of survival motor neuron (SMN) protein. In recent years, treatments able to restore SMN protein levels have been developed. However, these therapeutic strategies are not a definitive cure, and molecular mechanisms that lead to SMA are not completely understood. In this context, further studies to elucidate the molecular basis of the disease and the development of complementary strategies to regenerate damaged motor axons could play a pivotal role in ameliorating the outcome of SMA. In this regard, it has been recently demonstrated that the myelination of motor axons is impaired in SMA, and boosting neuregulins triggers the ensheathment of axons. This review discusses how a “preconditioning” phenomenon could trigger axonal terminal regeneration. We examine the “preconditioning stimuli” that can ameliorate this aspect of SMA pathology, which cannot be reversed by postnatal SMN protein induction.
Keywords: Spinal muscular atrophy, Neuromuscular junction regeneration, Schwann cells, Myelination
Highlights
This paper presents how sub-lethal stimuli could regenerate damaged motor axons, thus playing a pivotal role in ameliorating the outcome of SMA. In this regard, it has been recently demonstrated that the myelination of motor axons is impaired in SMA, and boosting neuregulins triggers the ensheathment of axons, which cannot be reversed by postnatal SMN protein induction.
Introduction
The neuromuscular junction (NMJ) is the anatomical structure where an electrical stimulus traveling along motor neuron (MN) axons is transformed into a chemical one, culminating in acetylcholine release from the pre-synaptic terminal. Acetylcholine released in the synaptic cleft binds its receptors located on muscle fibers, thus stimulating muscle contraction (Tintignac et al., 2015). In addition, NMJ consists of three pivotal elements: a) MNs axonal terminals, b) the muscle fibers, and c) a carpet of non-myelinating perisynaptic Schwann cells (PSCs) or teloglia (Reed et al., 2022). In particular, PCSs play a central role in forming, maintaining, remodeling, and regenerating the NMJ (Santosa et al., 2018). Interestingly, unlike the myelinating Schwann cells that wrap axons to establish efficient electrical impulse propagation, PSCs are not deeply characterized. Moreover, NMJ is the Achilles’ heel of several neurodegenerative diseases, such as spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS). In particular, it has been demonstrated that in these pathological conditions, NMJ cellular impairment occurs quite in advance of the first clinical symptoms (Voigt et al., 2010; Vinsant et al., 2013). After peripheral nerve trauma, native muscle reinnervation is triggered within a relatively narrow time window (approximately 12-18 months) (Boyd et al., 2011). To date, great efforts have been made to understand the molecular mechanisms that underlie the hastening of neural regeneration in neurons. However, the regeneration pathways that influence nerve repair are poorly investigated in NMJ, specifically in PSCs (Santosa et al., 2018). In particular, PSCs are an efficient tool to trigger axonal regeneration after denervation or nerve injury. Indeed, nerve terminals respond by growing or ‘sprouting’. This phenomenon is driven primarily by PSC activation (Son et al., 1996), which starts three days after motor nerve injury (Kang et al., 2014). Briefly, PSCs “perceive” neuron damage and extend cytoplasmic processes, thus forming “bridges” that allow rapid connections between denervated and innervated synaptic sites (Kang et al., 2014). Moreover, during the reinnervation, PSCs play a pivotal role in removing cellular debris through phagocytic activity, which is important in promoting successful nerve regeneration (Duregotti et al., 2015). Although PSC process extension has been well described in the literature (Son et al., 1996; Kang et al., 2014), the molecular mechanisms that trigger this phenomenon are still under investigation.
An overview of molecular mechanisms occurring in PSC during NMJ regeneration
Neuregulins-Erb-b2 receptor tyrosine kinase 2/Erb-b3 receptor tyrosine kinase 3 (ErbB2/ErbB3)
Neuregulins (NRGs) are a class of growth factors that show sequence similarities with epidermal growth factor (EGF). In particular, neuregulin 1 (NRG1) is encoded by the homonym gene that has different promoters that give rise to different protein isoforms through processes of alternative splicing (Falls, 2003). All NRG1 isoforms share an EGF-like domain that binds and activates ErbB receptors. Moreover, the expression pattern of Nrg1 isoforms is peculiar; for instance, NRG1 type III is mainly expressed by neuronal cells, and type I NRG1 by a few restricted neuronal cell types and by mesenchymal cells of many organs (Fleck et al., 2013). These two isoforms of Nrg1 (type III and type I) have different functions. NRG1 type III is involved in myelination, and Nrg type I is involved in muscle spindle induction (Birchmeier and Bennett, 2016). Furthermore, some NRGs are membrane-bound proteins that must be released from specific proteases to perform their functions. Conversely, other NRGs, after proteolytic cleavage, can remain bound to cellular surfaces. In both cases, the NRG receptors can detect signals from distant cells, directly from neighboring cells (paracrine signaling), or occasionally from the same origin cell, thus triggering autocrine signaling (Falls, 2003). NRG1 type I and type III arise from proteolytic processing dependent on beta-secretase 1 (Bace1) and members of the disintegrin and metalloproteinase (Adam) family (Fleck et al., 2013). Neuregulins possess a broad spectrum of functions like migration, cell fate decisions, morphogenesis, proliferation, and the control of cell size. They target many different cell types (glial cells, neurons, muscle, and epithelial cells) (Birchmeier and Bennett, 2016). In this review, we focus on the role of PSC in NMJ regeneration and repair. In particular, PSCs can respond to NRG stimuli through two main receptors: ErbB2 and ErbB3 (Meyer and Birchmeier, 1995). These receptors show peculiar characteristics since ErbB2 is a ligand-less receptor; by contrast, ErbB3 lacks tyrosine kinase activity (Woldeyesus et al., 1999). The formation of ErbB2 and ErbB3 heterodimers results in receptor tyrosine phosphorylation that activates signaling cascades (Citri et al., 2003) involving rat sarcoma virus (Ras)/ mitogen-activated protein kinase (MAPK)/extracellular signal-regulated protein kinase 1/2 (ERK1/2),phosphoinositide 3-kinase (PI3K), protein kinase B (Akt), focal adhesion kinase (FAK), and c-Jun N-terminal kinase (JNK) (Newbern and Birchmeier, 2010; Yarden and Sliwkowski, 2001). Interestingly, NRG1 type I and type III are crucial to repair and remyelination after nerve lesions in the peripheral nervous system, where different nerve injuries can occur, ranging from traumatic damage to NMJ impairments. In particular, Stassart et al. (2013) showed that NRG1 type I expression is transiently upregulated in PSCs after an injury, most likely due to the loss of axon contact and axonal NRG1 type III signaling (Figure 1). Moreover, developmental myelination seems strictly driven by the NRG1 type III isoform, while myelin repair results from the contribution of the molecular cascades activated by NRG1 type I and III isoforms (Birchmeier and Bennett, 2016).
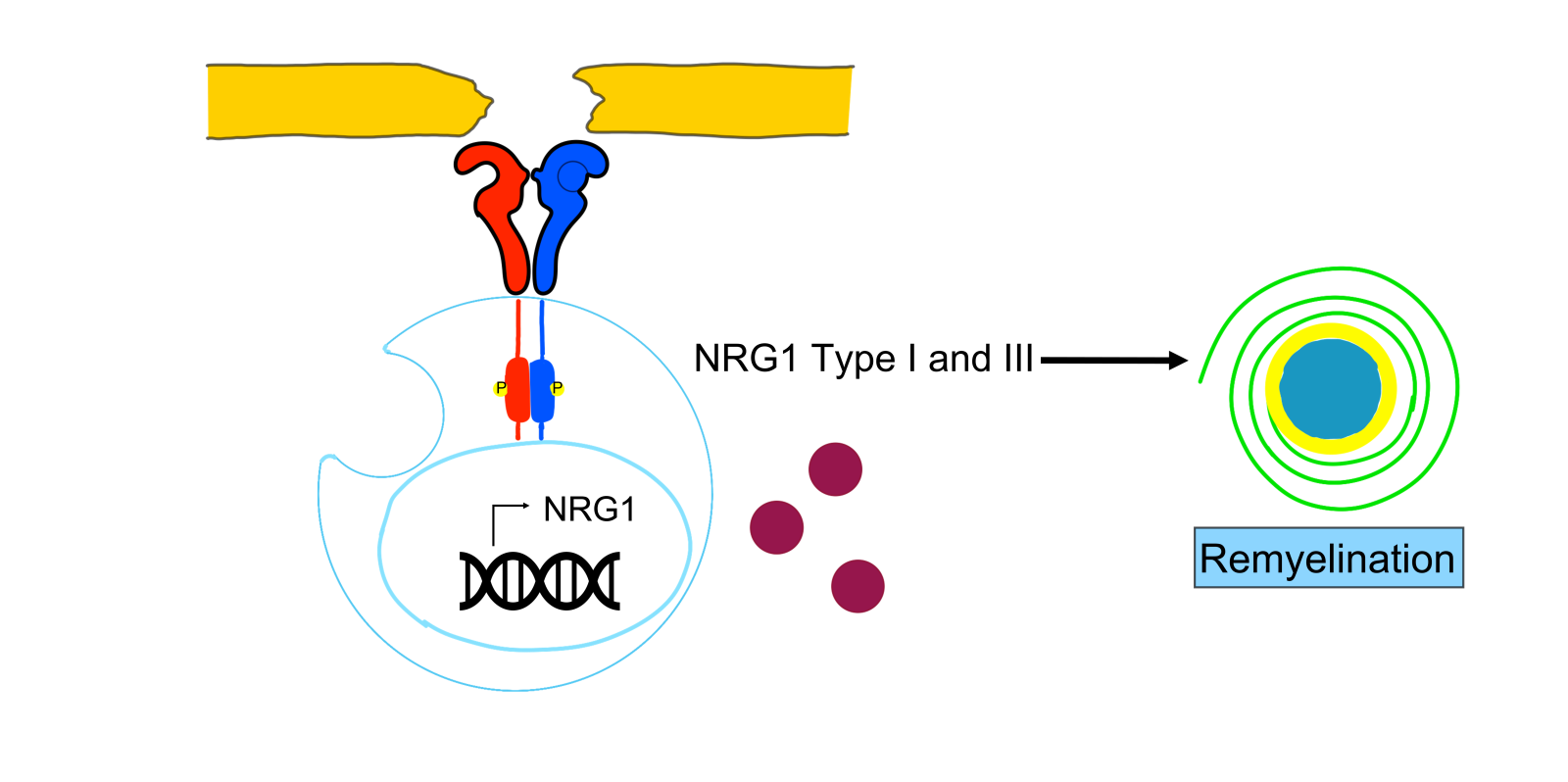
In a new window | Download PPT
Figure 1. In peripheral Schwann cells, NRG1 type I expression is transiently upregulated after a harmful injury. Loss of axonal contact triggers denervated Schwann cells to transiently express NRG1 as an autocrine/paracrine signal that promotes Schwann cell differentiation and remyelination. Moreover, developmental myelination seems strictly driven by the NRG1 type III isoform, while myelin repair results from the contribution of molecular cascades activated by NRG1 type I and III isoforms.
Role of sublethal stimuli in the regeneration of motorneuron axon terminals
The term preconditioning refers to a phenomenon in which sublethal stimuli trigger the activation of endogenous adaptive processes underlying tissue resistance to a subsequent and more severe injury (Pignataro et al., 2009). In particular, it has been shown that some animal-derived presynaptic neurotoxins (α-latrotoxin, β-bungarotoxin) and hydrogen peroxide (H2O2) represent “controlled” harmful stimuli that determine an acute and highly reproducible motor axon terminal degeneration followed by complete regeneration in peripheral nerve terminals (Duregotti et al., 2015). Moreover, the above-cited neurotoxins share a common injury mechanism that involves a large entry of calcium into axon terminals; calcium overload, in turn, triggers the release of molecules called “alarmins” composed of H2O2, cytochrome c (Cyt c), and mitochondrial DNA (mtDNA) from mitochondria (Duregotti et al., 2015)). Duregotti and colleagues (2015) demonstrated that H2O2, produced by degenerating nerve terminals after exposure to presynaptic neurotoxins, activates PSC involved in axon repair through pro-regenerative ERK signaling. Moreover, the acquisition of macrophagic-like activity of PSC, promoted by H2O2, plays a pivotal role in removing nerve cell debris and facilitating reinnervation (Duregotti et al., 2015). Indeed, the neutralization of H2O2 through catalase enzyme activity delays NMJ regeneration in vivo after toxin-induced damage and decreases ERK phosphorylation in PSC in culture. Additionally, Cyt c and mtDNA released from mitochondria can exit from damaged neurons in an unconventional manner, such as in secretory lysosomes and multivesicular body-derived exosomes (Frühbeis et al., 2012), where they then contribute to the activation of the ERK pathway in PSC (Duregotti et al., 2015). Intriguingly, H2O2 has been identified as a major signal of NMJ regeneration after an initial and reversible degeneration. Intriguingly, H2O2 can cross the membrane of PSCs and drives their remodelling by triggering profound changes in mRNA transcription and translation of genes involved in their morphological changes.
Moreover, the transcriptomic profile obtained from primary Schwann cells exposed to sublethal doses of H2O2 showed an upregulation in many classes of genes related to cytoskeleton remodeling and motility, including the annexin (Anxa) family (Negro et al., 2022). Importantly, H2O2 applied to the distal chamber of a microfluidic device works as a chemoattractant for primary Schwann cells that start to extrude cellular prolongations along its concentration gradient. In particular, Anxa2 appears to correlate with the edges of protrusions that Schwann cells extend when they receive signals from nearby axons (Poitelon et al., 2015). Moreover, Negro and colleagues (2022) demonstrated that Anxa2 mRNA localizes in Schwann cell projections, where it probably supports the initial stages of cytoskeletal changes that precede phagocytosis mediated by PSC (Mithal et al., 2012; Negro et al., 2022).
Furthermore, it has been well established that the chemokine stromal-derived factor 1 (SDF-1), also named CXCL12α, plays a pivotal role in brain development (Ma et al., 1998) and acts through its receptor, CXCR4. Interestingly, CXCR4 null mice show brain development abnormalities in the cerebellum, dentate gyrus, cerebral cortex, and dorsal root ganglia (DRG) (Mithal et al., 2012). In the brain cortex, SDF1/CXCR4 signaling from multiple neuronal cellular subtypes is used to reach proper migration (Stumm et al., 2003), and the CXCL12α/CXCR4 “axis” role seems to be crucial in axon guidance (Borrell and Marín, 2006). In spinal MNs, the NMJ degeneration/regeneration process is induced by α-latrotoxin and lasts five days. In particular, among the novel genes involved in axon regeneration, Negro and colleagues (2022) focused their attention on the role of the chemokine CXCL12α. Its expression in PSCs peaks four hours after hindlimb injection of α-latrotoxin. It has also been demonstrated that in primary cultured spinal cord MNs, CXCL12α produced by PSCs stimulates axon elongation through its receptor CXCR4, which is expressed at the growing tips of axons (Figure 2). Conversely, a CXCL12α-neutralizing antibody or a specific CXCR4 inhibitor drastically delays recovery from MNs degeneration in an in vivo model of α-latrotoxin insult (Negro et al., 2022).
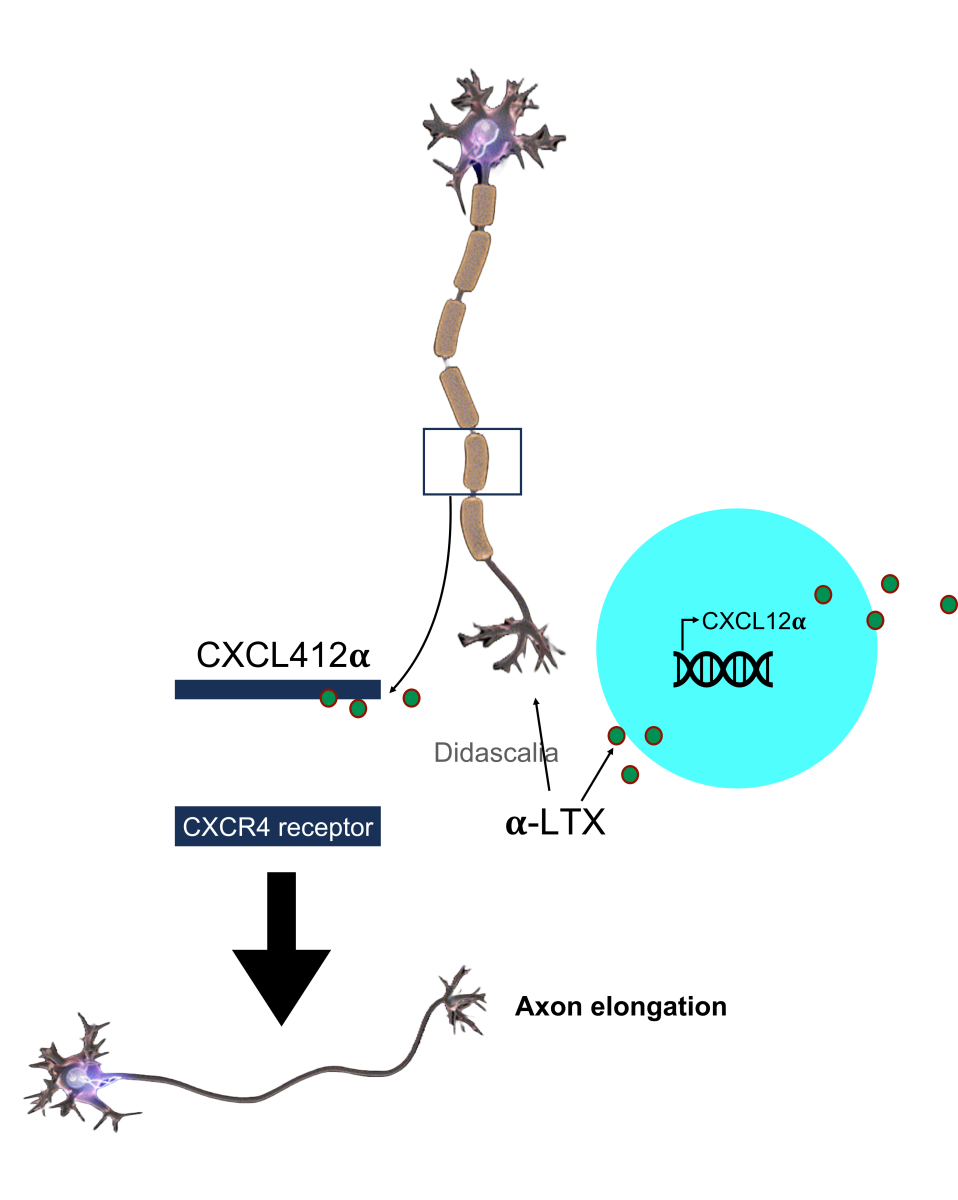
In a new window | Download PPT
Figure 2. Hindlimb injection of α-latrotoxin, increased the transcription of chemokine CXCL12α by PSCs. In primary cultured spinal cord motor neurons, CXCL12α stimulates axon elongation through CXCR4 interaction, whose expression is augmented at the growing tips of axons.
Further investigations showed that H2O2, produced in response to axon injury, can change the transcriptomic profile of genes involved in producing extracellular matrix components, such as connective tissue growth factor (CTGF). Indeed, CTGF upregulation in murine PSCs follows a biphasic model correlating with two temporal peaks of expression: the first one during the acute phase of NMJ degeneration and the second one during axon regeneration (Negro et al., 2022). The neutralization of CTGF, using a specific antibody, delays the regeneration of motor axon terminals and misregulates the migration of PSCs in the injury site. In contrast, the alarmin H2O2 promotes CTGF synthesis and release by primary PSCs favoring NMJ regeneration (Negro et al., 2022). Moreover, little is known about CTGF function. A study conducted on zebrafish showed that this growth factor supports glial proliferation and the subsequent formation of “new tissue” between two stumps to reconnect nerve transections, thus promoting spinal cord regeneration (Mokalled et al., 2016).
PSC role in the pathophysiology of spinal muscular atrophy
Spinal muscular atrophy (SMA) is the most common fatal motor neuron disease in children and is inherited in an autosomal recessive pattern. SMA is caused by the loss of the survival motor neuron (SMN) protein, and this disease is characterized by the progressive degeneration of the lower motor neurons of the anterior horn of the spinal cord, resulting in muscle weakness and atrophy, mainly affecting the lower extremities (Wirth et al., 2020). The SMN region, located on 5q13.2, has a very complex organization comprising repetitive sequences, deletions/duplications, and pseudogenes and contains two SMN genes in tandem, SMN1 (telomeric) and SMN2 (centromeric copy). Most humans have one or more copies of the SMN2 gene, which is absent in other species. Both genes share 99.9% of their sequence and can encode for a 294 amino acid full-length protein. However, while SMN1 encodes for a full-length 294 amino acid protein (SMN1), only a small percentage of SMN2 produces the full-length functional protein, and due to alternative splicing, it encodes for a shorter protein lacking exon 7 (SMN2). SMN2 is unstable and not fully functional, thus prone to degradation (Burghes and Beattie, 2009) (Figure 3).
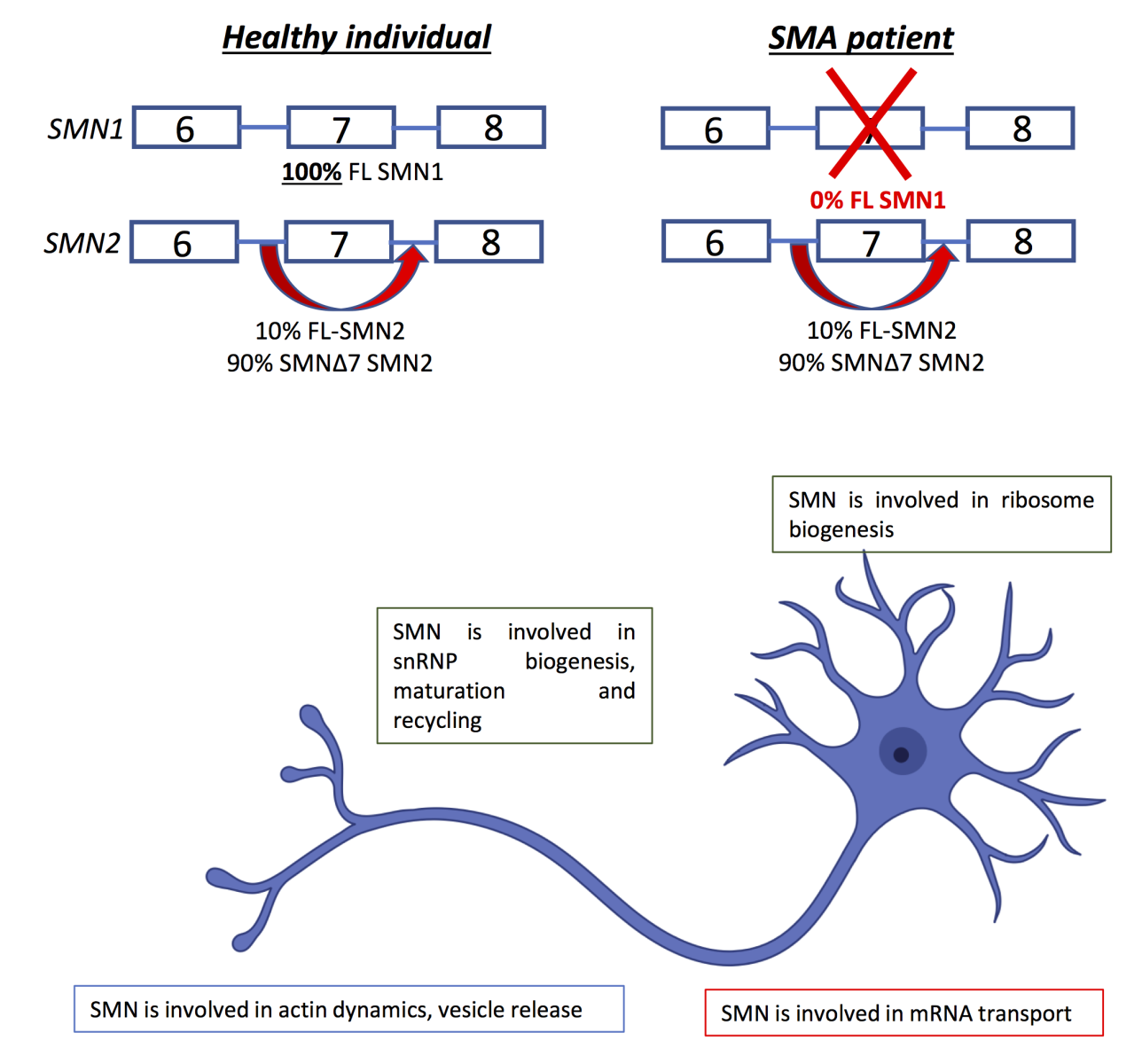
In a new window | Download PPT
Figure 3. The SMN1 gene and splicing events produce the normal full- length SMN1 protein and the SMN2 gene produces the Δ7 SMN2 protein by alternative splicing. The two genes differ by five nucleotides. The C840C>T variant at position +6, destroys an exonic splicing enhancer and instead creates an exonic splicing silencer in SMN2. This allele also produces a small amount (~10%) of FL SMN protein. Healthy individuals have two copies of SMN1 and two copies of the SMN2 genes. Deletion or gene conversion mechanisms lead to different amounts of SMN protein and different types of SMA and disease severity (Mokalled et al., 2016).
A homozygous deletion of the SMN1 gene accounts for 95% of SMA cases, and the rest is due to a small deletion, splicing, or missense mutation (Burghes and Beattie, 2009). Accordingly, the number of SMN2 copies directly correlates with disease severity and determines, together with the age of onset, four clinical groups (type 1-4), which inversely correlate with the number of SMN2 copies and SMN protein levels. SMN protein is ubiquitously expressed throughout the body and has an important role in transcriptional processes, protein translation, autophagy, and ubiquitination (Chaytow et al., 2018). Currently, there are three FDA-approved drugs for SMA: Nusinersen (Spinraza), adeno-associated virus serotype 9 (AVXS - 101) (Zolgensma), and risdiplam (Evrysdi), which are all SMN-enhancing therapies (Singh et al., 2020). However, all treatments have limitations, such as a narrow therapeutic window, sparse efficacy, and targeting limited to specific tissues (Lefebvre and Sarret, 2020). Moreover, they are not accessible to all patients in terms of costs and route of administration (Hamilton and Gillingwater, 2013). Furthermore, Schwann cells play a pivotal role in the formation of myelin sheath in MNs (Son et al., 1996) that are not correctly myelinated in models of severe SMA (Hunter et al., 2014). It has been reported that Schwann cells isolated from SMA mice were disabled to respond to differentiation signals and displayed an abnormal expression of key myelin formation proteins (Hunter et al., 2014). Interestingly, restoration of SMN in Schwann cells reversed myelination defects in MNs and improved neuromuscular function (Hunter et al., 2016). In particular, Hunter and colleagues (2014) performed experiments in co-cultures of PSCs (isolated from SMA mice) and healthy neurons from dorsal root ganglion (DRG). In this condition, PSCs negatively influenced the stability of neuronal wild-type cells, and this effect was not caused by the secretion of neurotoxic factors but rather by defects in the extracellular matrix composition of the Schwann cells. Moreover, a large variety of studies have shown a substantial delay in the development of motor axons and Schwann cells in SMA mice in utero (Kong et al., 2023), where individual motor axons must undergo a maturation process to become large, myelinated axons able to carry signals at rapid conduction velocities. The first and pivotal stage that occurs after Schwann cell proliferation is the maturation of individual motor axons with the sheathing by Schwann cells in a ratio of 1:1 (Kong et al., 2021). By contrast, SMA motor axons are grouped into clusters, which do not respect the 1:1 ratio of ensheathment due to reduced Schwann cell proliferation.
Furthermore, SMA motor axons, although sufficiently mature to be myelinated, are smaller in diameter and have a slow conduction velocity. These impairments precede neurodegeneration that occurs rapidly during the neonatal phase (Kong et al., 2021). Therefore, in ventral roots ganglia of SMA mice at embryonic and early postnatal time points, the axons show an immature morphology related to reduced myelination (Vinsant et al., 2013). In particular, NRG1 type III drives the ensheathment and myelination of motor axons during development (this isoform of NGR1 is produced by MNs in the developing spinal cord) (Kariyawasam et al., 2022) but also helps to preserve the functional integrity of peripheral nerve axons. A specific role of SMN deficiency on slowed axon development and degeneration has yet to be defined. However, NRG1 expression is reduced in the spinal cord and ventral root ganglia of severe SMA patients and in mouse models of SMA (Meyer et al., 1997), probably due to defective mRNA processing or transcription of the NRG1 gene (Meyer et al., 1997). Indeed, NRG1 transcripts undergo extensive alternative splicing, and SMN protein is a key factor for small nuclear ribonucleoproteins maintenance that is fundamental for spliceosome fidelity (Buonanno, 2010). Furthermore, it has been shown that overexpression of NRG1 in SMA mice not only improves axonal myelination but also trafficking and post-translational neurofilament modifications, which establish axonal radial diameter (Hsieh et al., 2011). Moreover, Kong and colleagues (2021) demonstrated that boosting NRG1 isoform III expression in SMA can increase axon diameters and ventral root ganglia size due to reinforced myelination caused by increased PSC number. NRG1 type III overexpression also improves neonatal motor axon conduction velocity and accelerates motor axon development, although it cannot prevent distal axon degeneration of the NMJ (Meyer et al., 1997).
Proteomic profiling studies in SMA mice revealed an impairment in the expression of molecules bound to cellular ubiquination homeostasis. (Kong et al., 2023). In particular, reduced expression of the protein ubiquitin-like modifier activating enzyme (Uba1) in the spinal cord and skeletal muscle of SMA has been reported, trigging the accumulation of β-catenin and myelination defects in a β-catenin independent manner. Indeed, using an inhibitor of Uba-1 in wild-type, Schwann cells reproduced the defective myelination phenotype observed in SMA. Specifically, in mice, the ensheathment of axons driven by Schwann cells starts at embryonic day 12.5 (E12.5), the first compact myelin sheaths appear at postnatal day 1 (P1), and the entire myelination process is completed around postnatal day 10 (P10). By contrast, this myelination process lasts longer in humans and ends in utero (Kariyawasam et al., 2022). Treatment of severe SMA mouse models with SMN C3 (Ando et al., 2020), an SMN splicing modifier (Buonanno, 2010) during gestation, improved SMA outcomes (Kong et al., 2021), including the ensheathment of axons, thus maximizing their maturation and the survival of MNs in severe SMA fetuses (Kong et al., 2021).
Concluding remarks
Although gene therapy approaches already exist to block the progression of SMA, all treatments available have limitations, such as narrow therapeutic window, sparse efficacy, and targeting limited to specific tissues. Therefore, investigating complementary strategies to regenerate damaged motor axons could play a pivotal role in fine-tuning strategies able to prevent neurodegeneration. In particular, other studies must be conducted not only to avoid neurodegeneration “in utero” but also to stimulate the regeneration of axon terminals after a “preconditioning” phenomenon in newborn mice that the alarmins and α-latrotoxin may trigger at sub-toxic doses. Overall, understanding the mechanisms triggered by alarmins, NRG1 type III (crucial for repair and remyelination after nerve lesions) may be relevant for identifying druggable targets against SMA. Since SMA has been recognized as a multi-system disorder (Hamilton and Gillingwater, 2013), general treatment should be considered. In conclusion, it would be necessary to investigate developmental pathways triggered by “preconditioning stimuli” to ameliorate aspects of disease pathology that cannot be reversed by postnatal SMN protein induction alone.
Conflict of interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Prof. Pignataro, who serves on the editorial board, did not participate in the review of this article at any level.
Acknowledgments
The project was also funded under the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3, Call for tender No. 341 of 15/03/2022 of Italian Ministry of University and Research (MUR) funded by the European Union, NextGenerationEU [Project title ‘A multiscale integrated approach to the study of the nervous system in health and disease’ (MNESYS); code PE0000006, CUP D93C22000930002, MUR Concession Decree No. 1553 of 11/10/2022].
References
Giusy Laudati1
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.
Valeria Valsecchi1
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.
Xhesika Kolici1,2
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.. 2School of Advanced Studies, Centre for Neuroscience, University of Camerino.
Giuseppe Pignataro1
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, University of Naples Federico II, Naples, Italy.
Corresponding author:
Giuseppe Pignataro
Email: giuseppe.pignataro@unina.it
In a new window | Download PPT
Figure 1. In peripheral Schwann cells, NRG1 type I expression is transiently upregulated after a harmful injury. Loss of axonal contact triggers denervated Schwann cells to transiently express NRG1 as an autocrine/paracrine signal that promotes Schwann cell differentiation and remyelination. Moreover, developmental myelination seems strictly driven by the NRG1 type III isoform, while myelin repair results from the contribution of molecular cascades activated by NRG1 type I and III isoforms.
In a new window | Download PPT
Figure 2. Hindlimb injection of α-latrotoxin, increased the transcription of chemokine CXCL12α by PSCs. In primary cultured spinal cord motor neurons, CXCL12α stimulates axon elongation through CXCR4 interaction, whose expression is augmented at the growing tips of axons.
In a new window | Download PPT
Figure 3. The SMN1 gene and splicing events produce the normal full- length SMN1 protein and the SMN2 gene produces the Δ7 SMN2 protein by alternative splicing. The two genes differ by five nucleotides. The C840C>T variant at position +6, destroys an exonic splicing enhancer and instead creates an exonic splicing silencer in SMN2. This allele also produces a small amount (~10%) of FL SMN protein. Healthy individuals have two copies of SMN1 and two copies of the SMN2 genes. Deletion or gene conversion mechanisms lead to different amounts of SMN protein and different types of SMA and disease severity (Mokalled et al., 2016).
Metrics
| Full-Text | Supporting Information | ||
|---|---|---|---|
| Number | 4955 | 27 | 0 |
Copyright © 2017 Conditioning Medicine, All Rights Reserved.
Address: Conditioning Medicine Editorial Office, 3500 Terrace Street, Pittsburgh, PA, 15213, USA